
The Dichotomous Role of N-methyl-D-Aspartate Receptors in Ischemic Stroke
Adam R. Holmes1, Francis J. Castellino1,2, Rashna D. Balsara1,2*
1W. M. Keck Center for Transgene Research, University of Notre Dame, Indiana 46556, USA
2Department of Chemistry and Biochemistry, University of Notre Dame, Indiana 46556, USA
Abstract
Ischemia-mediated glutamate elevation causes activation of the N-methyl-D-aspartate receptor (NMDAR) and consequent excitotoxicity. This triggers a cascade of pathological events, including aberrant NMDAR ion channel kinetics, large neuronal Ca2+ influx, and activation of pro-death signaling pathways. Previous studies have shown that functional outcomes of post-ischemia are influenced by the type of GluN2 subunit assembled in the NMDAR (GluN2A, GluN2B, GluN2C, or GluN2D), as well as its cellular location. GluN2A-containing synaptic NMDAR’s activate pro-survival pathways, whereas, activation of GluN2B-containing extrasynaptic NMDARs results in cell death. However, there is no consensus omnium on the individual role of the GluN2 subunits in ischemia. Published studies suggest that the GluN2A, GluN2B, and GluN2C subunits can promote either neuronal death or survival, depending on the experimental model employed and the CNS region investigated. In this mini-review, we aim to succinctly outline the mechanisms that underlie the dichotomous role of the NMDAR in ischemic stroke and possible NMDAR--directed therapeutic approaches.
Introduction
The N-methyl-D-aspartate receptor (NMDAR) is a ligand and voltage gated ionotropic neuroreceptor that is co--agonized by glutamate and glycine. Activation of this receptor allows influx of Ca2+, the result of which induces neuronal signaling events and propagation of action potentials. These phenomena are requisite for the long--term potentiation and depression of pathways that mediate higher--evel executive functions. The NMDAR is present on the post-synaptic membrane of neurons, although studies have also noted their presence in non-neuronal cells, including astrocytes and oligodendrocytes. At resting potentials, these ligand-gated channels are blocked by Mg2+. Interaction of the NMDAR with glutamate in the synaptic cleft results in depolarization of post-synaptic neurons that is accompanied by influx of Ca2+ and Na+, and efflux of K+1. However, during neuropathologies, an excess of glutamate is released from the synaptic bouton accompanied by a surfeit of post-synaptic Ca2+ influx, resulting in a multitude of unfavorable downstream processes that lead to neuronal death2. Thus, NMDAR’s play a critical role in not only maintaining neuronal-related physiology, resulting from synaptic plasticity, but also in pathophysiological diseases, including ischemic stroke, Alzheimer’s disease, Parkinson’s disease, and alcohol dependence3.
Expression, Structural, and Functional Properties of NMDAR
Understanding the various properties of the NMDAR is integral to defining its functional role under physiological and pathological conditions. The NMDAR is a heterotetrameric complex, and the composition of the functional ion channel is regulated developmentally, temporally, and spatially. The channel is composed of two glycine-binding GluN1 subunits and two glutamate-binding GluN2 subunits. The mandatory GluN1 subunit has eight isoforms (GluN1-1a to 4a and GluN1–1b to 4b) that arise from alternative splicing4. There are four different GluN2 subunits (GluN2A-D) that are products of independent genes5-7. Most commonly, the NMDARs found in the forebrain are composed of either two GluN2A or two GluN2B subunits. GluN2C-containing NMDARs are ample in the cerebellum, hippocampus, and amygdala. GluN2D subunits are mainly associated with NMDARs of the hippocampus, brain stem, diencephalon, and basal ganglia8. A GluN3 (A/B) subunit that binds to glycine and co-assembles with GluN1/GluN2 subunits has been more recently discovered9. It is well known that the GluN2 subunits impart unique biophysical, biochemical, pharmacological, and electrophysiological properties to the ion channel in accordance with their specific physiological roles10 (Fig. 1). NMDARs can also exist as triheteromers composed of GluN1 and two different types of GluN2 subunits, which possess functional and gating characteristics distinct from the diheteromeric NMDAR11,12 (Fig. 2).
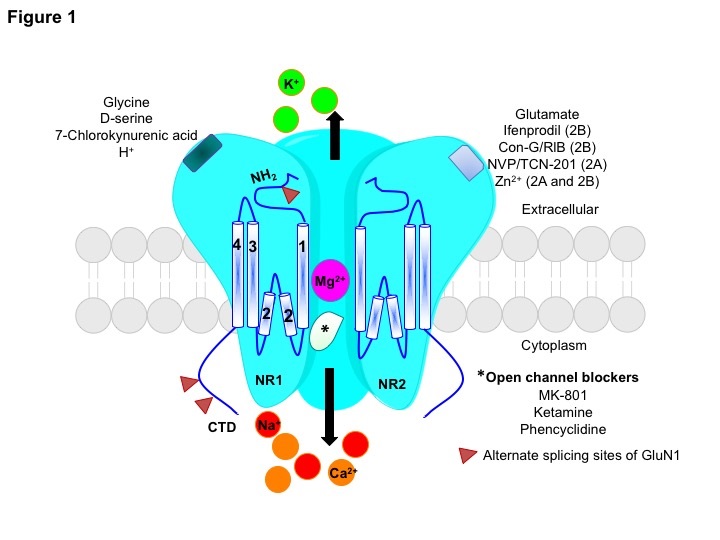
Figure 1: A schematic representation of the heterotetrameric NMDAR that consists of two GluN1 subunits and two GluN2(A/B) subunits. The GluN1 subunit can bind to glycine, D-serine, 7-Chlorokynurenic acid (antagonist), and extracellular protons (H+), and shows the splicing sites on exons 5, 21, and 22. The GluN2(A/B) subunit has the glutamate binding site, but can also bind to various pharmacological agents in a subunit-dependent manner. Ifenprodil and conantokin-G/RlB are selective antagonists for the GluN2B subunit, whereas, NVP-AAM077 and TCN-201 are GluN2A-specific antagonists. Extracellular Zn2+ binds to the GluN2A subunit with high affinity (nM) and to the GluN2B subunit with low affinity (µM). The TMD (1-4) topology that are connected by linker regions, the amino terminal end (NH2) and the C-terminal domain (CTD) are shown. Upon binding of the glutamate/glycine co-agonists, the Mg2+ block is removed allowing influx of Ca2+ and Na+ ions, and efflux of K+ ions. The open channel blockers MK-801, ketamine, and phencyclidine are some of the few uncompetitive NMDAR antagonists.
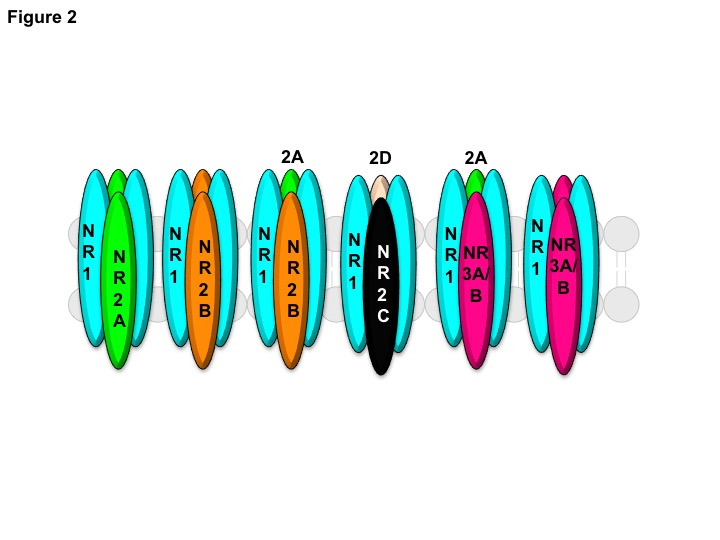
Figure 2: A schematic representation of various combinations of the NMDAR subunits present in the CNS. The functional NMDAR ion channel mostly exists as a diheteromer consisting of GluN1/GluN2A or GluN1/GluN2B subunits. Triheteromeric NMDARs can be made up of GluN1/GluN2A/GluN2B, GluN1/GluN2C/GluN2D, or GluN1/GluN2A/GluN3A/B subunits. The NMDAR ion channels consisting of GluN1/GluN3A/B subunits preferentially bind to glycine over glutamate.
Expression of the GluN subunits is spatially and temporally regulated. Although, expression of the ubiquitous GluN1 subunit begins at embryonic day 14 and is present throughout the CNS, the distribution pattern of its various isoforms changes depending on the region of the brain, and is based on the alternate splicing of exons 5, 21, and 22. In rodents, the GluN1-1 isoform is found concentrated in the cortex and hippocampus, whereas, the GluN1-4 subunit is predominantly found in the thalamus and cerebellum. The GluN1-1a isoform lacking exon 5 is found mainly in the dentate gyrus and CA1-3 layer of the hippocampus. The GluN1-1b isoform containing exon 5 encoded highly charged amino acids in the N-terminal region is concentrated in the CA3 hippocampal region. The GluN1-3 subunit is poorly expressed13,14. High levels of GluN2B and GluN2D are observed in rodent embryos, but within the first two weeks of birth to adulthood levels of the GluN2A subunit increases. Embryonically, the GluN2B subunit is prevalent in the brain, but after birth its distribution is restricted to regions of the forebrain. Expression of the GluN2C subunit is delayed starting at post-natal day 10-11, and is mostly confined to the cerebellum. Expression of the GluN2D subunit continues from the embryo to adulthood in the diencephalon, mesencephalon, and spinal cord15. Similarly, the GluN3 subunits have contrasting expression profiles. GluN3A expression is low in the embryonic stage that peaks at postnatal day 8 in the neocortex, CA1 region of the hippocampus, olfactory bulb, cerebellum amygdala, thalamus, and hypothalamus, and decreases in adulthood16. Whereas, GluN3B expression levels are low during early postnatal stages and increase with age till adulthood, and are ubiquitously distributed in the CNS17,18. Expression of the various subunits was found to be enhanced from midgestation to early childhood (20 – 400 postconceptional weeks) compared to adults in the human parieto-occipital white matter, with GluN2A levels peaking at midgestation, and GluN1 and GluN2B levels peaking at the preterm period. Levels of GluN1, GluN2B, GluN2C, GluN2D, and GluN3A were higher in the parieto-occipital gray matter during early postnatal period (20 – 400 postconceptional weeks) compared to adults. However, GluN2A levels were lower in this region compared to adults19.
The GluN subunits contain an extracellular amino-terminal domain (ATD), a ligand-binding domain (LBD), a transmembrane domain (TMD), and an intracellular C-terminal domain (CTD). The ATD allosterically regulates agonist potency by binding to Zn2+, ifenprodil, and polyamines, thereby influencing the gating properties of the NMDAR20,21. X-Ray crystallography studies of GluN1a/GluN2B-containing NMDARs suggests this allosteric regulation is due to tight packing of the ATD and LBD. The TMD of the GluN1a/GluN2B heterotetramer forms a pseudo-four-fold symmetry due to rearrangement of the M4 helices, an arrangement that aids in regulating the gating properties22. Additionally, binding of glutamate agonists and competitive antagonists is modulated by the LBDs23. The strategically placed helical segments of the TMD forms the channel pore that gates the voltage-dependent Mg2+ block, thereby regulating Ca2+ influx and electrophysiological conductance24,25. The cytoplasmic CTD is largely known for partnering with various signaling molecules and scaffolding proteins that form complexes involved in signaling and receptor trafficking. Additionally, the CTD of the GluN2A, GluN2B, and GluN2C subunits affects gating kinetics26. Truncated CTD of the GluN2A subunit caused reduced Ca2+ charge transfer and impaired spatial working memory in mice27. Structurally, the GluN3 subunits exhibit the same modular properties as the other GluN subunits, and bind to glycine or D-serine. Alternate splicing of the Grin3a mRNA gives rise to long (GluN3A-L) and a short (GluN3A-S) isoforms, whereas, five isoforms of the GluN3B subunit exist due to in-frame deletions or insertions at the N-terminal or C- terminal domains28, 29.
The NMDAR displays distinct slow activation and deactivation properties compared to other iGluRs, such as AMPA and kainite receptors30. Not only are the functional kinetics activated by binding of glutamate/glycine, but the NMDAR is also subject to modulation by endogenous Mg2+, H+, and Zn2+. The composition of the GluN2 subunits of the NMDAR governs its single channel conductance and Ca2+ permeability. Typically, GluN2A- and GluN2B-containing diheteromers manifest large conductances (~50 pS), longer channel open times (3-5 ms), and higher open probabilities (Po, ~0.4-0.5), when compared to GluN2C-or GluN2D-containing diheteromers that exhibit low conductances (~37 pS), shorter channel open times (0.5-1 ms), and lower open probabilities (Po, ~0.01)31. Also, GluN2A- and GluN2B- containing NMDAR channels are more sensitive to voltage-dependent Mg2+ inhibition compared to GluN2C- and GluN2D-containing NMDAR channels32,33. Moreover, Ca2+ influx is higher through channels containing GluN2A or GluN2B subunits compared to receptors that incorporate GluN2C or GluN2D subunits34,35. Mutagenesis studies have shown that TMD sites Ser632 and Leu657 mediate unique channel properties of GluN2A and GluN2D, respectively, that is dependent on the interaction of GluN2 and GluN1 subunits36. Crystal structure analysis showed that residues 658Asp-Arg-Pro-Glu-Glu-Arg663 in the linker region of LBD-TMD of GluN1 subunit is responsible for high permeation of Ca2+15.
The GluN3 subtypes bind to glycine/D-serine, and in particular, GluN3A confers a dominant negative function to the ion channel by countervailing the canonical synaptic and structural plasticity of GluN2-containing NMDARs by destabilizing synapses by tagging them for elimination. In this manner, GluN3A subunits fine-tunes synaptic connections promoting synaptic stabilization which is vital during postnatal development37. The GluN3 subunits form a functional triheteromeric complex with GluN1 and either GluN2A (in neurons and hippocampus), GluN2B (neurons), or GluN2C (in oligodendrocytes) subunits imparting biochemical/electrophysiological properties that are unique from the GluN2-containing receptors38-40. The diheteromeric GluN3 subunit-containing ion channels are refractory to blockage by Mg2+, AP-5, memantine, and MK-80141, but recently a series of small molecule compounds, TK13, TK30, and TK80 were found to specifically antagonize the GluN3 subunit42. The GluN3A-containing triheteromeric NMDAR channels have lower single-channel conductance of ~28 pS, lower open probability (Po, ~0.03), and low Ca2+ permeability43,44.
Discerning the Role of GluN2 Subunits in Stroke
Lack of glucose/oxygen results in loss of ionic homeostasis causing depolarization of neuronal cells resulting in accrual of pathological levels of glutamate. Ischemia also hinders clearance of glutamate resulting in glutamate spillover in the extrasynaptic spaces. Downstream, this ischemia-mediated excitotoxicity hyperactivates the NMDAR ion channel with an overload of cellular Ca2+ influx making it the underlying mechanism of ischemia-induced excitotoxicity45. Furthermore, perturbations in the ionic balance causes spreading depolarization and edema in the grey matter accompanied by depression of spontaneous activity also known as nonspreading silence, which can be negated by the NMDAR agonist, ketamine46. Acute deprivation of oxygen/glucose also induces NMDAR-evoked ischemic long-term potentiation (iLTP) that blocks physiological LTP and is thought to be the basis of aberrant synaptic plasticity that alters neuronal connectivity47. Post-ischemic LTP can be mitigated by application of ifenprodil, an GluN2B-specific antagonist48. The molecular outcome of ischemia excitotoxicity is neuronal necrosis or apoptosis due to calpain activation49, generation of reactive oxygen species50, and mitochondrial damage51. These ischemia-driven excitotoxic events are manifested into clinical symptoms of stroke.
The composition and interaction of the GluN1 subunit with the specific GluN2 subunit plays a principal role in regulating NMDAR-derived currents and Ca2+ influx, a process integral to maintaining physiological activity in the CNS. It has been well established that the NMDAR plays opposing roles in eliciting distinct signaling pathways depending on their cellular location. Ca2+/Calmodulin activates synaptically-located NMDAR’s, which transduces the PI3K/Akt pathway by serially complexing with various signaling molecules and subsequently promoting activation of pro-survival pathways (ERK1/2, CREB) and genes (BTG2, BCL6, BDNF)52-54. Inhibition of pro-apoptotic molecules (JNK, GSK3ß, BAD) also occurs55,56. Since the GluN2A subunits are abundantly found in the synaptic NMDAR channel, this subunit is associated with pro-survival pathways57. Conversely, GluN2B subunits are enriched in extrasynaptic NMDAR channels which partner with different proteins, such as PSD-95, DAPK1, PTEN, STEP, and SFK/Panx1, to form pro-death complexes during ischemia. These interactions lead to further upregulation of death target proteins, including calpains, MAPK/JNK, and SREBP1, within 1-12 hours of stroke onset, causing neuronal apoptosis58 (Fig. 3).
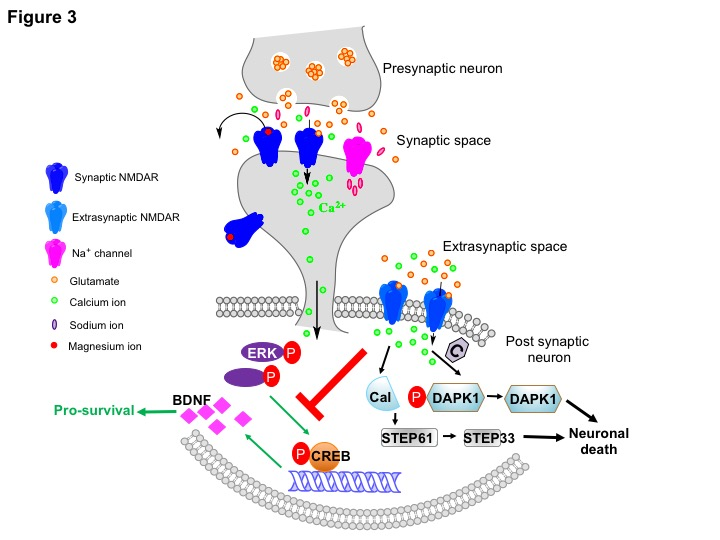
Figure 3: Signaling pathways activated by synaptic or extrasynaptic NMDARs. Physiological levels of Ca2+ influx due to stimulation of the NMDAR by glutamate at the synaptic cleft activates ERK signaling, which in turn phosphorylates CREB at Ser133 resulting in transcription of pro-survival molecules like Brain-derived Neurotrophic Factor (BDNF). During an ischemic event, glutamate spill over activates extrasynaptic NMDARs that inhibits the ERK/CREB signaling axis. Additionally, several pro-death pathways are also activated, of which, the death-associated protein kinase 1 (DAPK1)-and calpain (Cal)-mediated pathways are shown. Pathological levels of Ca2+ influx stimulates calcineurin (C) to dephosphorylate DAPK1 at Ser308 to promote excitotoxicity. Caplain mediates neuronal death by cleaving striatal-enriched protein tyrosine phoshatase (STEP) 61 to inactive STEP 33.
Since, ischemia-induced damage is multifactorial, several molecular events involving various signaling pathways occur during ischemia that contributes towards excitotoxicity. The PSD-95 scaffolding protein specifically interacts with GluN2B and neuronal nitric oxide synthase (nNOS) via its PDZ1/PDZ2 domains causing over activation of nNOS and hence increased levels of nitric oxide (NO)59. Pathological levels of NO precipitate a chain of protein oxidation and lipid peroxidation reactions eventually causing DNA fragmentation and neuronal death60. Disrupting the PSD-95/GluN2B interaction by using the Tat-NR2B9c or NA-1 peptide successfully attenuated infarct size with improvement in neurological deficits in rat and non-human primate models of stroke61-63. Another repercussion of pathological Ca2+ neuronal influx is the activation of death-associated protein kinase 1 (DAPK) protein that binds to the cytoplasmic tail of GluN2B thereby stimulating the pro-apoptotic activity of DAPK1 by the Ca2+/calmodulin/calcinerin phosphatase axis. Disrupting the GluN2B/DAPK1 interaction with the Tat-GluN2BCT1292-1304 peptide mitigated the excitotoxicity due to ischemia64. The Akt signaling molecule is also negatively affected by ischemia-induced Ca2+ influx due to interaction of the upstream phosphatase and tensin homolog deleted on chromosome 10 (PTEN) molecule with GluN1/GluN2B-containing extrasynaptic ion channels.
The direct interaction between GluN1/PTEN promotes ischemia-mediated neuronal death, whereas down regulating PTEN levels reduced neuronal injury in vitro and in vivo by increasing phosphorylation of Akt and BAD forging pro-survival activity and decreasing neuronal death65. Also, reduction of PTEN levels decreased NMDAR-evoked extrasynaptic current, but not synaptic activity of the NMDAR ion channels, thus obliquely regulating GluN2B activity65. Uncoupling the PTEN/NMDAR interaction by peptide Tat-K13 abrogated ischemia-induced nuclear accumulation of PTEN and conferred neuroprotection with improved neurological deficits66.
A thorough understanding of the role of individual GluN2 subunits in ischemia is lacking due to the differential roles played by the GluN2A and GluN2B subunits during ischemia, as well as due to the extensive presence of GluN1/GluN2A/GluN2B triheteromeric NMDAR’s in the adult forebrain. Mice deficient in the GluN2A subunit showed abrogated ischemia-induced brain injury compared to WT mice. In contrast, selectively blocking the GluN2A subunit with the glutamate antagonist NVP-AAM077 resulted in a larger infarct size, indicating that GluN2A was essential for ischemic neuroprotection in vivo67. The pro-apoptotic effect of GluN2A was attributed to phosphorylation of Ser1232 by cyclin-dependent kinase in a model of ischemia, whereas, decreased tyrosine phosphorylation was associated with decreased ischemia-induced neuronal death68.
Inhibition of the GluN2B subunit with specific antagonists has proved to be beneficial in animal models of stroke. It has been demonstrated that administration of GluN2B-specific inhibitors, ifenprodil and conantokin-G, ameliorates ischemia-mediated infarct size and neuronal death by decreasing neurological deficits, restoring cytoarchitecture, and increasing the expression of pro-survival CREB molecules69,70. When extrasynaptic, GluN2B-containing NMDAR channels were treated with conantokin-G, increased neuronal survival was observed by increased pro-survival levels of ERK, CREB, and enhanced mitochondrial viability, compared to a non-subunit-specific conantokin-T71. Perturbing the interaction of the GluN2B CTD with PSD95, CMKIIα, and clathrin adaptor protein-2 mitigated the detrimental effects of O2/glucose deprivation of cultured cortical neurons72. Although, the GluN2B subunit plays a prominent role in cell death induced by ischemia, it is not involved in cell death caused by cardiac arrest and cardiopulmonary resuscitation73. Post-ischemia, mRNA expression levels of the GluN subunits changes temporally causing ischemic-mediated apoptosis. Within the first 24 hours following stroke, mRNA levels of GluN1 increases, but decreases by day 7. Whereas, the mRNA levels of GluN2A and GluN2B decreases initially, but start to increase at 48 hours post- ischemia74.
The role of the anatomically restricted GluN2C subunit in ischemia is ambiguous. GluN2C plays a neuroprotective role in a murine model of global cerebral ischemia in the hippocampus due to upregulation of the GluN2C subunit and decreased Ca2+ influx75. This contrasts with another study that reported that a deficiency of GluN2C subunit in mice conferred neuroprotection, as observed by smaller infarct size and less cerebral edema in a middle carotid artery occlusion model of ischemia, compared to WT mice76. Furthermore, it has been shown that post-ischemic GluN2C-/- mice exhibit enhanced neurological recovery associated with less cytoarchitectural deficits and Tauopathy, as well as decreased levels of Fyn kinase and diminished phosphorylation of Tyr1336 of the GluN2B subunit in the cerebral cortex compared to WT mice77. The direct role of GluN2D in ischemia has not as yet been elucidated. The neonates and adults of naked mole rats have an unusually high proportion of GluN2D subunits that impedes Ca2+ influx, thus rendering these animals highly tolerant to living in hypoxic conditions78.
NMDAR channels containing the GluN3A subunits have low Ca2+ permeability and sensitivity to Mg2+, and play a protective role during ischemic insult, both in vivo and in vitro. The GluN3A subunit dampens ischemia-mediated excitotoxicity by counteracting the hyperactivation of GluN2A- and GluN2B-containing ion channels. GluN3A-/- mice showed impaired neurological recovery, larger infarct volume, and elevated cell death compared to WT mice79.
NMDAR as a Therapeutic Target of Ischemia
Although occurrence of ischemic stroke is prevalent (~800,000 strokes annually in the USA), treatment plans available to counter the detrimental effects of stroke are limited, especially at the neurovascular and neuroprotective level. Administration of the thrombolytic agent, alteplase (recombinant tissue type-Plasminogen Activator) is the most widely used drug to dissolve the clot and restore blood flow. However, the effectiveness of alteplase is limited to the narrow window of ~3.5 hours after onset of stroke during which it should be administrated, and it could also increase the risk of hemorrhage80. Another option of removing the clot is by mechanical embolectomy that can be performed as late as 8-12 hours after onset of stroke81. Progression of stroke is linear and the NMDAR-driven excitotoxic effects of stroke leading to neuronal death is a relentless process. Therefore, the optimal plan for stroke therapy would primarily involve removal of clot followed by secondary neuroprotective strategies. To this end several preclinical studies had developed inhibitors that specifically targeted the NMDAR.
Although blockade of the NMDAR by various antagonists was effective in attenuating ischemic damage in murine and rodent models, the clinical availability of NMDAR-directed antagonists remains elusive. Some non-competitive NMDAR inhibitors, such as, dizocipline, selfotel, aptiganel hydrochloride, and gavistinel, were investigated but suspended at various stages of clinical trials because of adverse side-effects that included psychotic and/or neurological complications82. The GluN2B-specific inhibitor class, ifenprodil and its analogues, were well-tolerated in clinical trials, yet were not effective compared to placebo administrations83. Treatment with various neuroprotective compounds, e.g., MgSO4 and glycine-site inhibitors, have also proved to be ineffective. Conantokins, which are neuroactive peptides derived from the Conus species of marine snails, which effectively inhibit NMDAR-evoked currents and Ca2+ influx in a subunit-specific manner, have been evaluated for epilepsy and pain, but globally affect other receptors84.
Several groups have reported that memantine which is a non-competitive antagonist of the NMDAR used to treat Alzheimer’s disease was also found to be efficacious for treatment of stroke. Improvement in neurological deficits accompanied by diminished astrogliosis and enhanced vascular density in the penumbra region of the core infarct was observed in mice treated with memantine administered after induction of stroke. However, this recovery was non-neuroprotective as reduction in core infarct size was not observed, although increase in brain derived neurotrophic factor and phospho-tyrosine was observed in the peri-infarct area indicating memantine-stimulated improvement in plasticity85. Prophylactic administration of memantine at a low dosage of 0.2 mg/kg significantly decreased infarct size with improved behavioral outcomes, but administration of higher doses of memantine (2-10 mg/kg) was counterproductive and potentiated ischemic injury in mice86. Memantine is a well-tolerated drug widely used to treat patients with Alzheimer’s disease, and pre-clinical research data supports the feasibility of memantine being utilized to treat patients with stroke. Lipton and colleagues synthesized analogs of memantine, the NitroMemantines that proved to be significantly more neuroprotective in treating ischemia compared to memantine by effectively blocking hyperactivation of extrasynaptic NMDARs in a voltage-dependent manner and allowing sustained synaptic activity87. Thus, making memantine and its analogs attractive therapeutic candidates for treatment of ischemia. In fact, early phase 1 clinical trials are in progress to evaluate the efficacy of memantine in patients with acute ischemic stroke.
Recent emphasis has been placed on development of short peptides that inhibit interaction of the CTD of GluN2 subunits with pro-death signaling pathways. The NA-1 peptide that perturbs the GluN2B/PSD-95/nNOS-directed synthesis of nitric oxide is undergoing phase II clinical trials88. Additionally, inhibiting nNOS expression also mitigates glutamate-mediated death-signaling MAPK p38 activation, preventing ischemia-induced neuronal death89. Some other peptides that negate death signaling pathways downstream of the NMDAR, and confer neuroprotection post-ictus are the D-JNK-1 and Tat-INDIP peptides that interfere with the stress-associated JNK and SREBP1 transcription factor respectively90,91. Another set of reagents that have efficacious neuroprotective properties are the polyarginine rich peptides (R12, R15, R18 and R18D) which are able to decrease infarct volume and improve stroke-mediated neurological deficits92,93. These cationic arginine-rich peptides (CARP) decrease glutamate-induced excitotoxicity and neuronal Ca2+ influx by decreasing surface expression of GluN2B, activate pro-survival pathways, and preserve neuronal mitochondrial integrity with minimum in vivo toxicity94,95. However, for neuroprotective agents to be successful, it is essential that the penumbra region that surrounds the core infarct which has reduced cerebral blood flow (10-35%), but has energy reserves and synaptic activity is redeemable96. Combination of mechanical clot removal and treatment with neuroprotective drugs would freeze the penumbra and prevent it from becoming part of the infarct core if ischemia is left untreated. In fact, current investigative/clinical emphasis is on boosting neural repair in the penumbra by increasing blood flow by collateral circulation97. Despite the high risk and costs associated with clinical trials, the pharmaceutical industry is pursuing the development of neuroprotective drugs due to the major health and economic burden that stroke has on the aging population.
Conclusions
It is clear that the NMDAR plays a key role in the pathology of ischemic stroke. However, there is conflicting literature regarding the role of the different GluN2 subunits in ischemia. These opposing results may stem from post-ischemic changes in expression and activity of the various GluN subunits in a spatio-temporal dependent manner. The type of ischemia model employed, and the duration of the insult, also influences the expression and activity of GluN2 subunits. An ideal neuroprotective agent is one that would selectively block the deleterious NMDAR-driven pathways without globally affecting normal synaptic function.
References
- Dingledine R, Borges K, Bowie D, et al. The glutamate receptor ion channels. Pharmacol Rev. 1999; 51: 7-61.
- Weilinger NL, Maslieieva V, Bialecki J, et al. Ionotropic receptors and ion channels in ischemic neuronal death and dysfunction. Acta Pharmacol Sin. 2013; 34: 39-48.
- Loftis JM, Janowsky A. The N-¬? methyl-¬? D-¬? aspartate receptor subunit NR2B: localization, functional properties, regulation, and clinical implications. Pharmacol Ther. 2003; 97: 55-85.
- Sugihara H, Moriyoshi K, Ishii T, et al. Structures and properties of seven isoforms of the NMDA receptor generated by alternative splicing. Biochem Biophys Res Commun. 1992; 185: 826-832.
- Anson LC, Chen PE, Wyllie DJ, et al. Identification of amino acid residues of the NR2A subunit that control glutamate potency in recombinant NR1/NR2A NMDA receptors. J Neurosci. 1998; 18: 581-589.
- Laube B, Hirai H, Sturgess M, et al. Molecular determinants of agonist discrimination by NMDA receptor subunits: analysis of the glutamate binding site on the NR2B subunit. Neuron. 1997; 18: 493-503.
- Ikeda K, Nagasawa M, Mori H, et al. Cloning and expression of the epsilon 4 subunit of the NMDA receptor channel. FEBS Lett. 1992; 313: 34-38.
- von Engelhardt J, Bocklisch C, Tönges L, et al. GluN2D-containing NMDA receptors-mediate synaptic currents in hippocampal interneurons and pyramidal cells in juvenile mice. Front Cell Neurosci. 2015; 9: 95.
- Nishi M, Hinds H, Lu HP, et al. Motoneuron-specific expression of NR3B, a novel NMDA-type glutamate receptor subunit that works in a dominant-negative manner. J Neurosci. 2001; 21: RC185.
- Traynelis SF, Wollmuth LP, McBain CJ, et al. Glutamate receptor ion channels: structure, regulation, and function. Pharmacol Rev. 2010; 62: 405-496.
- Tovar KR, McGinley MJ, Westbrook GL. Triheteromeric NMDA receptors at hippocampal synapses. J Neurosci. 2013; 33: 9150-9160.
- Cheriyan J, Balsara RD, Hansen KB, et al. Pharmacology of triheteromeric N-Methyl-D-Aspartate Receptors. Neurosci Lett. 2016; 617: 240-246.
- 13. Laurie DJ, Seeburg PH. Regional and developmental heterogeneity in splicing of the rat brain NMDAR1 mRNA. J Neurosci. 1994; 14: 3180-3194.
- Paupard MC, Friedman LK, Zukin RS. Developmental regulation and cell-specific expression of N-methyl-D-aspartate receptor splice variants in rat hippocampus. Neuroscience. 1997; 79: 399-409.
- Monyer H, Burnashev N, Laurie DJ, et al. Developmental and regional expression in the rat brain and functional properties of four NMDA receptors. Neuron. 1994; 12: 529-540.
- Pachernegg S, Strutz-Seebohm N, Hollmann M. GluN3 subunit-containing NMDA receptors: not just one-trick ponies. Trends Neurosci. 2012; 35: 240-249.
- Henson MA, Roberts AC, Pérez-Otaño I, et al. Influence of the NR3A subunit on NMDA receptor functions. Prog Neurobiol. 2010; 91: 23-37.
- Low CM, Wee KS. New insights into the not-so-new NR3 subunits of N-methyl-D-aspartate receptor: localization, structure, and function. Mol Pharmacol. 2010; 78: 1-11.
- Jantzie LL, Talos DM, Jackson MC, et al. Developmental expression of N-methyl-D-aspartate (NMDA) receptor subunits in human white and gray matter: potential mechanism of increased vulnerability in the immature brain. Cereb Cortex. 2015; 25: 482-495.
- Yuan H, Hansen KB, Vance KM, et al. Control of NMDA receptor function by the NR2 subunit amino-terminal domain. J Neurosci. 2009; 29: 12045-12058.
- Tian M, Ye S. Allosteric regulation in NMDA receptors revealed by the genetically encoded photo-cross-linkers. Sci Rep. 2016; 6: 34751.
- Karakas E, Furukawa H. Crystal structure of a heterotetrameric NMDA receptor ion channel. Science. 2014; 344: 992-997.
- Chen PE, Wyllie DJ. Pharmacological insights obtained from structure-¬? function studies of ionotropic glutamate receptors. Br J Pharmacol. 2006; 147: 839-853.
- Burnashev N, Schoepfer R, Monyer H, et al. Control by asparagine residues of calcium permeability and magnesium blockade in the NMDA receptor. Science. 1992; 257: 1415-1419.
- Sakurada K, Masu M, Nakanishi S. Alteration of Ca2+ permeability and sensitivity to Mg2+ and channel blockers by a single amino acid substitution in the N-methyl-D-aspartate receptor. J Biol Chem. 1993; 268: 410-415.
- Punnakkal P, Jendritza P, Kohr G. Influence of the intracellular GluN2 C-terminal domain on NMDA receptor function. Neuropharmacology. 2012; 62: 1985-1992.
- Bannerman DM, Niewoehner B, Lyon L, et al. NMDA receptor subunit NR2A is required for rapidly acquired spatial working memory but not incremental spatial reference memory. J Neurosci. 2008; 28: 3623-3630.
- Sun L, Margolis FL, Shipley MT, et al. Identification of a long variant of mRNA encoding the NR3 subunit of the NMDA receptor: its regional distribution and developmental expression in the rat brain. FEBS Lett. 1998; 441: 392-396.
- Domingues AM, Neugebauer KM, Fern R. Identification of four functional NR3B isoforms in developing white matter reveals unexpected diversity among glutamate receptors. J Neurochem. 2011; 117: 449-460.
- Forsythe ID, Westbrook GL. Slow excitatory postsynaptic currents mediated by N-methyl-D-aspartate receptors on cultured mouse central neurons. J Physiol. 1988; 396: 515-533.
- Paoletti P. Molecular basis of NMDA receptor functional diversity. Eur J Neurosci. 2011; 33: 1351-1365.
- Qian A, Buller AL, Johnson JW. NR2 subunit-¬? dependence of NMDA receptor channel block by external Mg2+. J Physiol. 2005; 562: 319-331.
- Qian A, Johnson JW. Permeant ion effects on external Mg2+ block of NR1/2D NMDA receptors. J Neurosci. 2006; 26: 10899-10910.
- Burnashev N, Zhou Z, Neher E, et al. Fractional calcium currents through recombinant GluR channels of the NMDA, AMPA and kainate receptor subtypes. J Physiol. 1995; 485 ( Pt 2): 403-418.
- Schneggenburger R. Simultaneous measurement of Ca2+ influx and reversal potentials in recombinant N-methyl-D-aspartate receptor channels. Biophys J. 1996; 70: 2165-2174.
- Siegler Retchless B, Gao W, Johnson JW. A single GluN2 subunit residue controls NMDA receptor channel properties via intersubunit interaction. Nat Neurosci. 2012; 15: 406-413, S401-402.
- Das S, Sasaki YF, Rothe T, et al. Increased NMDA current and spine density in mice lacking the NMDA receptor subunit NR3A. Nature. 1998; 393: 377-381.
- Perez-Otano I, Larsen RS, Wesseling JF. Emerging roles of GluN3-containing NMDA receptors in the CNS. Nat Rev Neurosci. 2016; 17: 623-635.
- Roberts AC1, Díez-García J, Rodriguiz RM, et al. Downregulation of NR3A-containing NMDARs is required for synapse maturation and memory consolidation. Neuron. 2009; 63: 342-356.
- Henson MA, Larsen RS, Lawson SN, et al. Genetic deletion of NR3A accelerates glutamatergic synapse maturation. PLoS One. 2012; 7: e42327.
- Chatterton JE, Awobuluyi M, Premkumar LS, et al. Excitatory glycine receptors containing the NR3 family of NMDA receptor subunits. Nature. 2002; 415: 793-798.
- Kvist T, Greenwood JR, Hansen KB, et al. Structure-based discovery of antagonists for GluN3-containing N-methyl-D-aspartate receptors. Neuropharmacology. 2013; 75: 324-336.
- Perez-Otano I, Schulteis CT, Contractor A, et al. Assembly with the NR1 subunit is required for surface expression of NR3A-containing NMDA receptors. J Neurosci. 2001; 21: 1228-1237.
- Sasaki YF, Rothe T, Premkumar LS, et al. Characterization and comparison of the NR3A subunit of the NMDA receptor in recombinant systems and primary cortical neurons. J Neurophysiol. 2002; 87: 2052-2063.
- Choi DW. Glutamate neurotoxicity in cortical cell culture is calcium dependent. Neurosci Lett. 1985; 58: 293-297.
- Sakowitz OW, Kiening KL, Krajewski KL, et al. Preliminary evidence that ketamine inhibits spreading depolarizations in acute human brain injury. Stroke. 2009; 40: e519-522.
- Stein ES, Itsekson-Hayosh Z, Aronovich A, et al. Thrombin induces ischemic LTP (iLTP): implications for synaptic plasticity in the acute phase of ischemic stroke. Sci Rep. 2015; 5: 7912.
- Picconi B, Tortiglione A, Barone I, et al. NR2B subunit exerts a critical role in postischemic synaptic plasticity. Stroke. 2006; 37: 1895-1901.
- Xu J, Kurup P, Zhang Y, et al. Extrasynaptic NMDA receptors couple preferentially to excitotoxicity via calpain-mediated cleavage of STEP. J Neurosci. 2009; 29: 9330-9343.
- Eliasson MJ, Huang Z, Ferrante RJ, et al. Neuronal nitric oxide synthase activation and peroxynitrite formation in ischemic stroke linked to neural damage. J Neurosci. 1999; 19: 5910-5918.
- Fujimura M, Morita-Fujimura Y, Murakami K, et al. Cytosolic redistribution of cytochrome c after transient focal cerebral ischemia in rats. J Cereb Blood Flow Metab. 1998; 18: 1239-1247.
- Joyal JL, Burks DJ, Pons S, et al. Calmodulin activates phosphatidylinositol 3-kinase. J Biol Chem. 1997; 272: 28183-28186.
- Hardingham GE, Fukunaga Y, Bading H. Extrasynaptic NMDARs oppose synaptic NMDARs by triggering CREB shut-off and cell death pathways. Nat Neurosci. 2002; 5: 405-414.
- Ivanov A, Pellegrino C, Rama S, et al. Opposing role of synaptic and extrasynaptic NMDA receptors in regulation of the extracellular signal-¬? regulated kinases (ERK) activity in cultured rat hippocampal neurons. J Physiol. 2006; 572: 789-798.
- Downward J. How BAD phosphorylation is good for survival. Nat Cell Biol. 1999; 1: E33-35.
- Yamaguchi A, Tamatani M, Matsuzaki H, et al. Akt activation protects hippocampal neurons from apoptosis by inhibiting transcriptional activity of p53. J Biol Chem. 2001; 276: 5256-5264.
- Liu Y, Wong TP, Aarts M, et al. NMDA receptor subunits have differential roles in mediating excitotoxic neuronal death both in vitro and in vivo. J Neurosci. 2007; 27: 2846-2857.
- Wu QJ, Tymianski M. Targeting NMDA receptors in stroke: new hope in neuroprotection. Mol Brain. 2018; 11: 15.
- Sattler R, Xiong Z, Lu WY, et al. Specific coupling of NMDA receptor activation to nitric oxide neurotoxicity by PSD-95 protein. Science. 1999; 284: 1845-1848.
- Lipton SA, Choi YB, Pan ZH, et al. A redox-based mechanism for the neuroprotective and neurodestructive effects of nitric oxide and related nitroso-compounds. Nature. 1993; 364: 626-632.
- Aarts M, Liu Y, Liu L, et al. Treatment of ischemic brain damage by perturbing NMDA receptor-PSD-95 protein interactions. Science. 2002; 298: 846-850.
- Bråtane BT, Cui H, Cook DJ, et al. Neuroprotection by freezing ischemic penumbra evolution without cerebral blood flow augmentation with a postsynaptic density-95 protein inhibitor. Stroke. 2011; 42: 3265-3270.
- Cook DJ, Teves L, Tymianski M. Treatment of stroke with a PSD-95 inhibitor in the gyrencephalic primate brain. Nature. 2012; 483: 213-217.
- Tu W, Xu X, Peng L, et al. DAPK1 interaction with NMDA receptor NR2B subunits mediates brain damage in stroke. Cell. 2010; 140: 222-234.
- Ning K, Pei L, Liao M, et al. Dual neuroprotective signaling mediated by downregulating two distinct phosphatase activities of PTEN. J Neurosci. 2004; 24: 4052-4060.
- Zhang S, Taghibiglou C, Girling K, et al. Critical role of increased PTEN nuclear translocation in excitotoxic and ischemic neuronal injuries. J Neurosci. 2013; 33: 7997-8008.
- Chen M, Lu TJ, Chen XJ, et al. Differential roles of NMDA receptor subtypes in ischemic neuronal cell death and ischemic tolerance. Stroke. 2008; 39: 3042-3048.
- Wang J, Liu S, Fu Y, et al. Cdk5 activation induces hippocampal CA1 cell death by directly phosphorylating NMDA receptors. Nat Neurosci. 2003; 6: 1039-1047.
- Ba?kaya MK, Rao AM, Donaldson D, et al. Protective effects of ifenprodil on ischemic injury size, blood-brain barrier breakdown, and edema formation in focal cerebral ischemia. Neurosurgery. 1997; 40: 364-370; discussion 370-361.
- Balsara R, Dang A, Donahue DL, et al. Conantokin-G attenuates detrimental effects of NMDAR hyperactivity in an ischemic rat model of stroke. PLoS One. 2015; 10: e0122840.
- Balsara R, Li N, Weber-Adrian D, et al. Opposing action of conantokin-G on synaptically and extrasynaptically-activated NMDA receptors. Neuropharmacology. 2012; 62: 2227-2238.
- Vieira MM, Schmidt J, Ferreira JS, et al. Multiple domains in the C-terminus of NMDA receptor GluN2B subunit contribute to neuronal death following in vitro ischemia. Neurobiol Dis. 2016; 89: 223-234.
- Quillinan N, Grewal H, Deng G, et al. Region-specific role for GluN2B-containing NMDA receptors in injury to Purkinje cells and CA1 neurons following global cerebral ischemia. Neuroscience. 2015; 284: 555-565.
- Liu Z, Zhao W, Xu T, et al. Alterations of NMDA receptor subunits NR1, NR2A and NR2B mRNA expression and their relationship to apoptosis following transient forebrain ischemia. Brain Res. 2010; 1361: 133-139.
- Chung C, Marson JD, Zhang QG, et al. Neuroprotection Mediated through GluN2C-Containing N-methyl-aspartate (NMDA) Receptors Following Ischemia. Sci Rep. 2016; 6: 37033.
- Kadotani H, Namura S, Katsuura G, et al. Attenuation of focal cerebral infarct in mice lacking NMDA receptor subunit NR2C. Neuroreport. 1998; 9: 471-475.
- Holmes A, Zhou N, Donahue DL, et al. A deficiency of the GluN2C subunit of the N-methyl-D-aspartate receptor is neuroprotective in a mouse model of ischemic stroke. Biochem Biophys Res Commun. 2018; 495: 136-144.
- Peterson BL, Park TJ, Larson J. Adult naked mole-rat brain retains the NMDA receptor subunit GluN2D associated with hypoxia tolerance in neonatal mammals. Neurosci Lett. 2012; 506: 342-345.
- Lee JH, Wei ZZ, Chen D, et al. A neuroprotective role of the NMDA receptor subunit GluN3A (NR3A) in ischemic stroke of the adult mouse. Am J Physiol Cell Physiol. 2015; 308: C570-577.
- National Institute of Neurological D, Stroke rt PASSG. Tissue plasminogen activator for acute ischemic stroke. N Engl J Med. 1995; 333: 1581-1587.
- Raychev R, Saver JL. Mechanical thrombectomy devices for treatment of stroke. Neurol Clin Pract. 2012; 2: 231-235.
- Hoyte L, Barber PA, Buchan AM, et al. The rise and fall of NMDA antagonists for ischemic stroke. Curr Mol Med. 2004; 4: 131-136.
- Gogas K.R. Glutamate-based therapeutic approaches: NR2B receptor antagonists. Curr Opin Pharmacol. 2006; 6: 68-74.
- Malmberg AB, Gilbert H, McCabe RT, et al. Powerful antinociceptive effects of the cone snail venom-derived subtype-selective NMDA receptor antagonists conantokins G and T. Pain. 2003; 101: 109-116.
- Lopez-Valdes HE, Clarkson AN, Ao Y, et al. Memantine enhances recovery from stroke. Stroke. 2014; 45: 2093-2100.
- Trotman M, Vermehren P, Gibson CL, et al. The dichotomy of memantine treatment for ischemic stroke: dose-dependent protective and detrimental effects. J Cereb Blood Flow Metab. 2015; 35: 230-239.
- Takahashi H, Xia P, Cui J, et al. Pharmacologically targeted NMDA receptor antagonism by NitroMemantine for cerebrovascular disease. Sci Rep. 2015; 5: 14781.
- Hill MD, Martin RH, Mikulis D, et al. Safety and efficacy of NA-1 in patients with iatrogenic stroke after endovascular aneurysm repair (ENACT): a phase 2, randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2012; 11: 942-950.
- Soriano FX, Martel MA, Papadia S, et al. Specific targeting of pro-death NMDA receptor signals with differing reliance on the NR2B PDZ ligand. J Neurosci. 2008; 28: 10696-10710.
- Borsello T, Clarke PG, Hirt L, et al. A peptide inhibitor of c-Jun N-terminal kinase protects against excitotoxicity and cerebral ischemia. Nat Med. 2003; 9: 1180-1186.
- Taghibiglou C1, Martin HG, Lai TW, et al. Role of NMDA receptor-dependent activation of SREBP1 in excitotoxic and ischemic neuronal injuries. Nat Med. 2009; 15: 1399-1406.
- Milani D, Knuckey NW, Anderton RS, et al. The R18 Polyarginine Peptide Is More Effective Than the TAT-NR2B9c (NA-1) Peptide When Administered 60 Minutes after Permanent Middle Cerebral Artery Occlusion in the Rat. Stroke Res Treat. 2016; 2016: 2372710.
- Milani D, Bakeberg MC, Cross JL, et al. Comparison of neuroprotective efficacy of poly-arginine R18 and R18D (D-enantiomer) peptides following permanent middle cerebral artery occlusion in the Wistar rat and in vitro toxicity studies. PLoS One. 2018; 13: e0193884.
- MacDougall G, Anderton RS, Edwards AB, et al. The Neuroprotective Peptide Poly-Arginine-12 (R12) Reduces Cell Surface Levels of NMDA NR2B Receptor Subunit in Cortical Neurons; Investigation into the Involvement of Endocytic Mechanisms. J Mol Neurosci. 2017; 61: 235-246.
- Edwards AB, Anderton RS, Knuckey NW, et al. Characterisation of neuroprotective efficacy of modified poly-arginine-9 (R9) peptides using a neuronal glutamic acid excitotoxicity model. Mol Cell Biochem. 2017; 426: 75-85.
- Astrup J, Siesjo BK, Symon L. Thresholds in cerebral ischemia -¬? the ischemic penumbra. Stroke. 1981; 12: 723-725.
- Regenhardt RW, Das AS, Stapleton CJ, et al. Blood Pressure and Penumbral Sustenance in Stroke from Large Vessel Occlusion. Front Neurol. 2017; 8: 317.