
Metabolic Modulation to Treat Cardiac Diseases: Role for Membrane Substrate Transporters
Jan F.C. Glatz*, Joost J.F.P. Luiken, Miranda Nabben
Department of Genetics & Cell Biology, Faculty of Health, Medicine and Life Sciences, Maastricht University, Netherlands
Abstract
There is growing recognition of the importance and multiple roles of substrate energy metabolism in both cardiac health and disease. Cardiac diseases are frequently accompanied by altered myocardial metabolism, while chronic changes in the type of myocardial substrate utilization are found to elicit cardiac contractile dysfunction. Examples are the increased glucose utilization, at the expense of fatty acids, in cardiac hypertrophy and ischemic heart failure, and the increased fatty acid utilization, at the expense of glucose, in obesity and diabetes-related cardiac dysfunction. Modulation of cardiac metabolism has emerged as a suitable therapeutic intervention in cardiac disease. Insights obtained during the past decade have revealed sarcolemmal substrate transport, facilitated by CD36 for fatty acids and by GLUT4 for glucose, to represent the main rate-governing kinetic step of substrate utilization, over-ruling intracellular sites of flux regulation. This suggests that manipulating the presence of substrate transporters in the sarcolemma may be an effective approach for metabolic modulation therapy. The present mini-review provides a short summary of the functioning of substrate transporters CD36 and GLUT4 in the heart, and discusses their application as targets for metabolic intervention.
Abbreviations
CD36: Cluster of Differentiation 36
FABPc: Cytoplasmic Fatty Acid-Binding Protein
FABPpm: Plasma Membrane Fatty Acid-Binding Protein
FATP: Fatty Acid Transport Protein
GLUT4: Glucose Transporter-4
SR-B2: Scavenger Receptor B2
VAMP: Vesicle-Associated Membrane Protein
v-ATPase: vacuolar-type H+-ATPase
Introduction
Cardiac energy metabolism is increasingly being recognized to play multiple roles in the cardiovascular system. Specifically, the unimpeded functioning of cardiac metabolism appears an important prerequisite for proper myocardial functioning. Consequently, metabolic changes that cause a progressive impairment of high-energy phosphate production often lead to heart failure. For instance, metabolic changes that occur in obesity and diabetes directly affect the cardiac contractile function and may explain why cardiovascular disease is the main cause of morbidity and mortality in patients with diabetes1,2. In general, heart failure – defined as a myocardial derangement causing systolic and/or diastolic ventricular dysfunction – is commonly associated with metabolic alterations, especially with regard to substrate preference, whereby distinct metabolic signatures are observed depending on the cause of heart failure (e.g. pressure-overload versus hypoxia/ischemia)2,3.
Evidence is accumulating that modulation of cardiac metabolism is a useful therapeutic approach in cardiac disease4. Such so-called ‘metabolic modulation’ intervention is directed towards counteracting the energy deprivation commonly seen in heart failure and towards re-installing the flexibility of the heart to adapt its energy provision to environmental influences (referred to as metabolic flexibility). Specific agents have been described that optimize cardiac substrate metabolism without exerting negative haemodynamic effects. These include metabolic agents stimulating or inhibiting fatty acid β-oxidation such as L-carnitine, etomoxir, perhexiline and trimetazidine, and agents stimulating glucose utilization such as dichloroacetate4-8. Importantly, while these metabolic modulators have shown promising results in experimental animal studies and some clinical trials4, other trials did not reveal any significant benefit or, in the case of etomoxir, even showed unacceptable side-effects that caused early termination of the trial9. It should be realized however, that these various agents are directed towards intracellular metabolic processing of a single type of substrate rather than securing the intracellular availability of a proper mixture of substrates that is needed for optimal energy provision10,11. Indeed, modulating such a mixture of substrates, for instance, through manipulating the entry of metabolic substrates into cardiomyocytes, is emerging as a novel therapeutic approach.
Recent insight into the regulation of myocardial substrate metabolism has revealed a pivotal role for membrane substrate transporters. The utilization of long-chain fatty acids, which form the predominant substrate for myocardial energy provision, is primarily determined by the rate of myocellular fatty acid uptake as facilitated by the presence of the fatty acid transporter CD36 (SR-B2) in the sarcolemma, and overrules the regulation of mitochondrial oxidation of fatty acids by the enzyme carnitine palmitoyl-transferase-1 (CPT-1)12. This molecular mechanism fully resembles the regulation of myocardial utilization of the other major substrate, i.e., glucose, which is dependent on the sarcolemmal presence of glucose transporters (GLUT1 and GLUT4)13. Importantly, both CD36 and GLUT4 have been implicated in dysregulated cardiac metabolism in pathophysiological conditions such as high-fat diet induced insulin resistance/diabetic cardiomyopathy and pressure-overload induced heart failure. These recent observations suggest that membrane substrate transporters may be preferable targets for metabolic modulation therapy to treat cardiovascular diseases.
In the present mini-review, we will explain the role of membrane substrate transporters in cardiac metabolism and contractile function in more detail, and examine their application as targets to treat cardiovascular diseases.
Myocardial Fatty Acid Uptake
The uptake of fatty acids into cardiomyocytes is facilitated by membrane-associated proteins with a predominant role for the transmembrane glycoprotein cluster-of-differentiation 36 (CD36; officially known as scavenger receptor SR-B2), which functions in cooperation with plasma membrane fatty acid-binding protein (FABPpm). The extracellular loop of CD36 contains a hydrophobic binding pocket that operates as an acceptor of fatty acids to promote the partitioning of the fatty acids and their delivery to the outer leaflet of the lipid bilayer (Fig. 1). Thereafter, the fatty acids ‘flip–flop’ to the inner leaflet of the membrane, which process occurs very fast and does not need assistance from membrane proteins12. The final step in the transmembrane transportation process is the desorption of the fatty acids from the inner leaflet and their binding to the cytoplasmic fatty acid carrier FABPc, which transfer is assumed to occur in specific membrane domains designated ‘lipid rafts’. The desorption is regarded as the rate-limiting step of overall transmembrane transport. CD36 is considered to facilitate this step by providing a docking site for FABPc or for acyl-CoA synthetases resulting in a highly efficient uptake process.
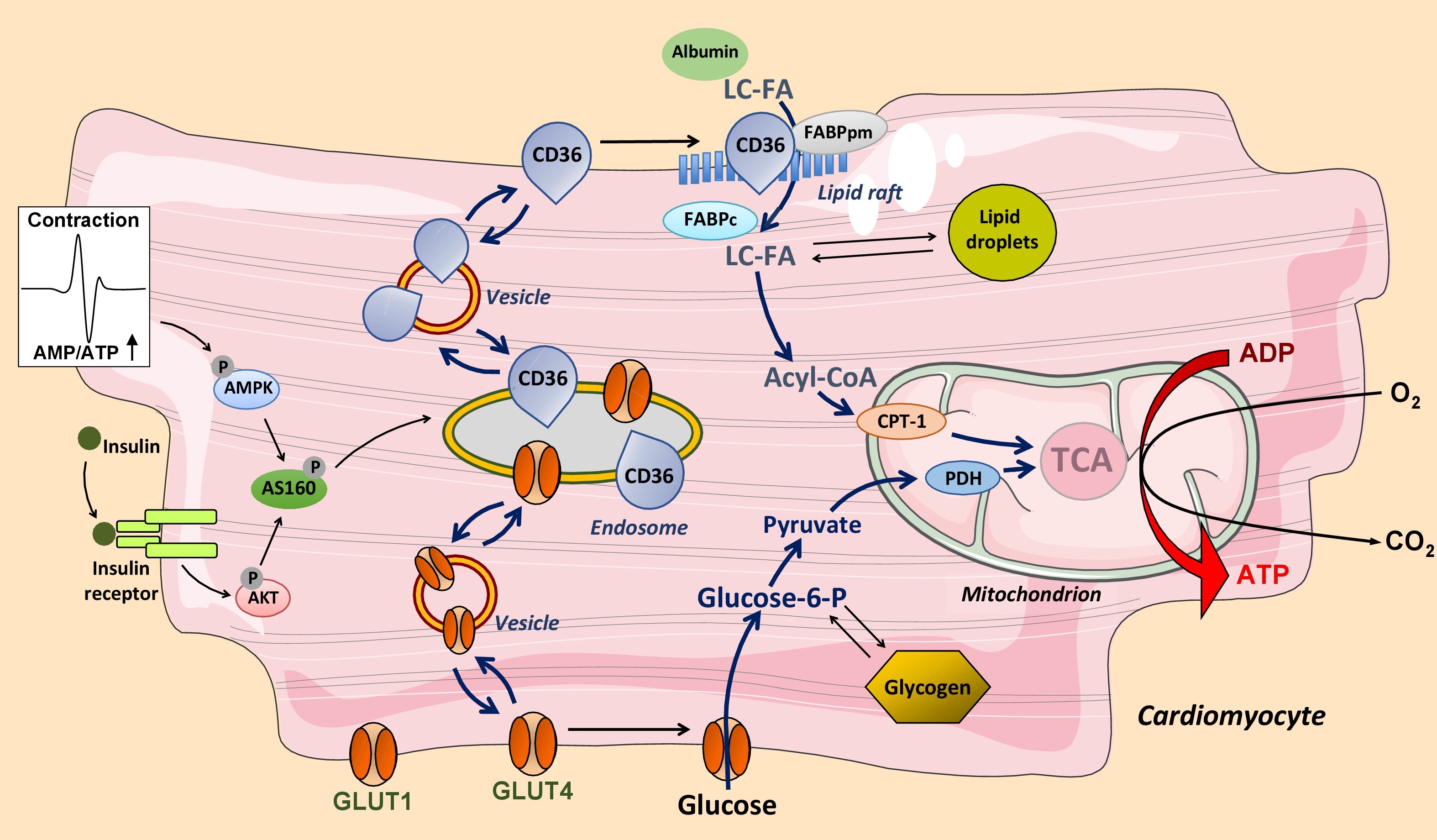
Figure 1: Regulation of long-chain fatty acid (LC-FA) and glucose utilization by cardiac myocytes. LC-FA uptake is mediated by fatty acid transporter CD36 (scavenger receptor (SR)-B2) and glucose uptake by glucose transporters GLUT1 and GLUT4. During increased contractile activity (mediated by AMP-activated kinase, AMPK) or in response to insulin both CD36 and GLUT4 reversibly translocate from an endosomal storage compartment to the sarcolemma to increase fatty acid and glucose uptake, respectively. The glucose transporter GLUT1 is constitutively expressed in the sarcolemma and contributes up to 25% of glucose uptake. Mitochondrial oxidation of LC-FA is controlled by the activity of carnitine palmitoyltransferase-1 (CPT-1) and that of glucose by pyruvate dehydrogenase (PDH). TCA, tricarboxylic acid cycle.
Under basal conditions, about half of the total cellular amount of CD36 is present in an intracellular storage depot, i.e., endosomes, from where CD36 can be recruited to translocate to the sarcolemma by vesicular transport. Either an increase in cardiomyocyte contraction or the presence of insulin each trigger, within minutes, the reversible translocation of CD36 from endosomes to the sarcolemma, which is paralleled by a proportional increase in the rate of cellular fatty acid uptake (Fig. 1)12,14. Taken together, CD36 is not merely a facilitator of transmembrane fatty acid transport, but through its reversible recycling between endosomes and the sarcolemma also serves a pivotal role as regulator of the rate of myocellular fatty acid uptake. It has been estimated that about 70% of myocardial fatty acid uptake is mediated and regulated by CD3615,16.
Control of the overall rate of myocardial fatty acid utilization is classically viewed to be governed by the activity of the mitochondrial enzyme, carnitine palmitoyl-transferase-1 (CPT-1)17. However, more recent work has questioned such a role for CPT-1 as it was found that the rate of fatty acid uptake and oxidation is not affected by partial inhibition of CPT-1 activity, yet is directly dependent on the amount of CD36 present in the sarcolemma18-21. As a result, while CPT-1 acts as a regulatory site for mitochondrial β-oxidation and merely serves a permissive role in overall fatty acid utilization, CD36-mediated trans-sarcolemmal transport appears the primary site of regulation of myocellular fatty acid flux12.
Myocardial Glucose Uptake
Glucose uptake into cardiomyocytes is mediated by two glucose transporters, i.e., GLUT1 and GLUT413,22, while members of the solute carrier family 2A (SLC2A) are not involved in the heart23. GLUT1 is constitutively present in the sarcolemma and mediates basal glucose uptake. GLUT4 is responsible for stimulus-inducible glucose uptake. In the adult heart, GLUT4 is expressed to a substantially higher extent than GLUT1, making the inducible glucose uptake component the quantitatively most important one13,22. Thus, it has been estimated that GLUT4 contributes to the rate of glucose uptake for at least 75%, or even more depending on cardiac workload13.
Substrate Transporters and Cardiac Disease
The corollary is that cardiac fatty acid and glucose utilization are determined largely by the sarcolemmal presence of CD36 and that of GLUT4, respectively. As a consequence, in cases of cardiovascular diseases associated with metabolic alterations, the sarcolemmal presence or functioning of these substrate transporters is altered as well. Numerous examples underline this notion.
Chronic oversupply of fatty acids to the heart, as occurs during high-fat diet consumption and in obesity, triggers a shift in myocardial energy provision towards an increased utilization of fatty acids at the expense of glucose1,24. This substrate switch eventually leads to the accumulation of specific lipid species in cardiomyocytes, followed by mitochondrial dysfunction, insulin resistance, and impaired contractile function, together referred to as diabetic cardiomyopathy25-27. Interestingly, detailed time-course studies have revealed that this order of events is initiated by a net translocation of CD36 from endosomes to the sarcolemma, which then leads to a concomitant increase in the rate of fatty acid uptake28,29. In vivo, this subcellular CD36 redistribution occurs rapidly (within days) while the other metabolic changes (e.g. mitochondrial dysfunction) are evident weeks later28. The pivotal early role of CD36 in this cascade of events suggests that manipulation of the sarcolemmal presence or activity of CD36 should prevent and/or regress high fat diet-induced toxic lipid accumulation and contractile dysfunction. Indeed, CD36-null mice are protected against high fat diet-induced loss of cardiac function30,31. In addition, cardiac-specific overexpression of nuclear receptor PPARα in mice resulting in enhanced cardiac fatty acid utilization and lipotoxic cardiomyopathy could be rescued by deletion of CD3632. Finally, isolated rat cardiomyocytes cultured in a high-palmitate containing medium show lipid accumulation, insulin resistance and a marked loss of contractile function, which all can be prevented by the addition of anti-CD36 antibodies to the culture medium33.
An increased utilization of glucose at the expense of fatty acids is seen during pressure overload-induced cardiac hypertrophy and is accompanied by an increased presence of GLUT4 at the sarcolemma34. Subjecting mice with transaortic constriction-induced cardiac hypertrophy30 or mice with genetically induced cardiac hypertrophy35 to a dietary intervention with a high fat-containing diet, in each case elicited normalization of glucose utilization (due to increased fatty acid utilization) together with the recuperation of contractile function. In both examples, it can be inferred that GLUT4 was redistributed leading to a net relocation towards intracellular stores.
Substrate Transporters as Targets for Intervention
Given the role of membrane substrate transporters in the pathogenesis of the cardiometabolic disease, they may be exploited as suitable targets for therapy with the aim to specifically alter either myocardial fatty acid or glucose utilization (Fig. 2). However, targeting their function at the sarcolemma by specific antibodies or inhibitors may not be a preferable systemic approach because such interventions would most likely also impair substrate uptake in other organs. Therefore, a preferable approach is to modulate the subcellular recycling of CD36 and/or that of GLUT4, aiming at manipulating their presence at the sarcolemma, and do so, if possible, specifically in the heart. This would require detailed knowledge of the factors involved in the vesicular trafficking machinery of CD36 and GLUT4 recycling between endosomes and the sarcolemma, and of the triggers that regulate the recycling process. During the last decade, there has been a marked expansion of knowledge in this field, in such a way that the concept of specific modulation of CD36 or GLUT4 recycling in a cardiac-specific manner seems feasible.
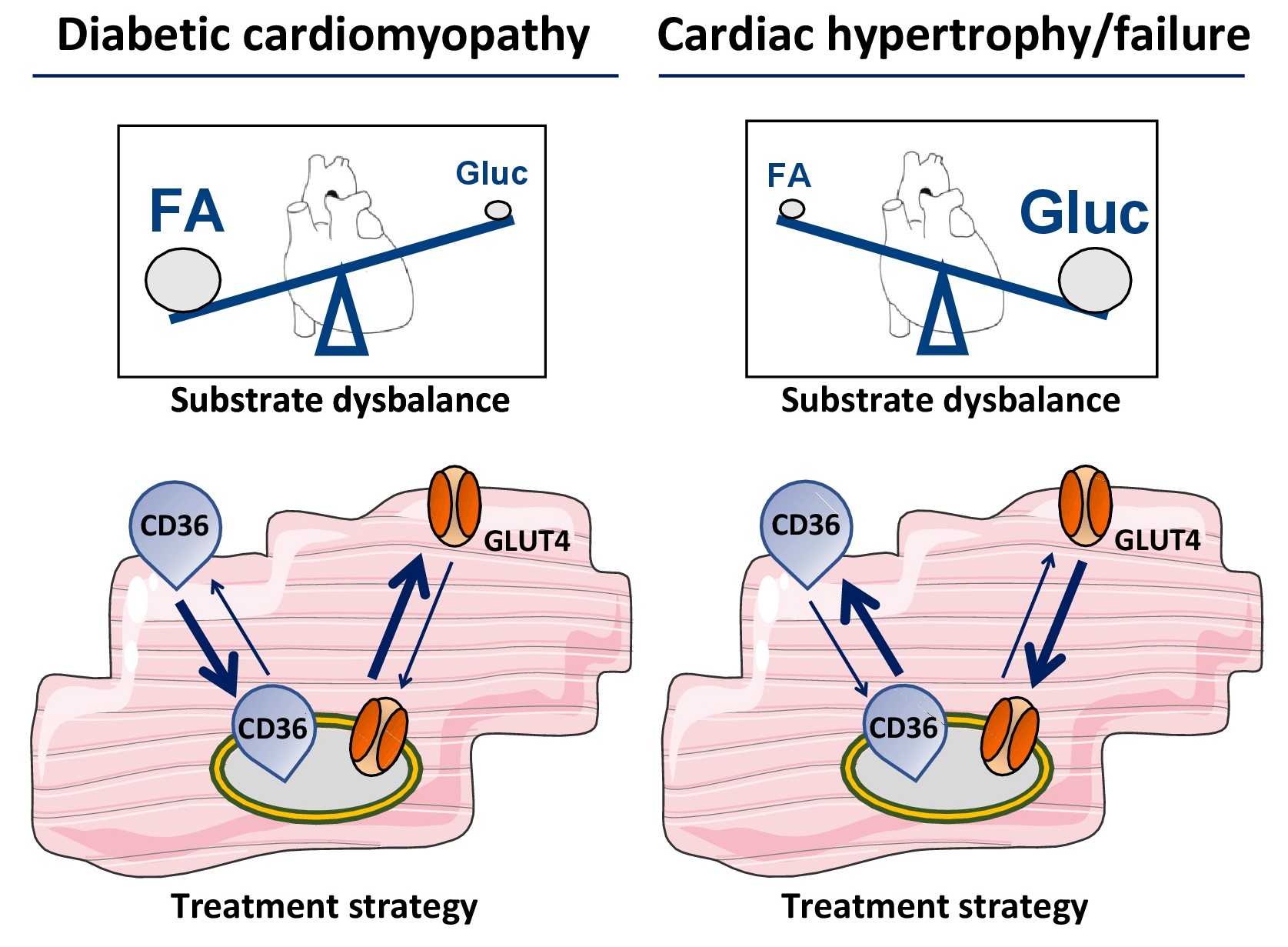
Figure 2: Scheme illustrating the application of substrate transporters for metabolic modulation therapy. Two examples are given. Left panels: In case of diabetic cardiomyopathy, cardiac substrate utilization is shifted towards increased utilization of long-chain fatty acids (FA) at the expense of glucose (Gluc). Interventions aimed at net internalization of CD36 and/or net translocation of GLUT4 to the sarcolemma (bold arrows) is expected to normalize substrate utilization and improve contractile function. Right panels: In case of hypertrophy and heart failure, cardiac substrate utilization is shifted in the other direction, i.e., towards increased utilization of glucose at the expense of fatty acids. In this case, interventions aimed at net internalization of GLUT4 and/or net translocation of CD36 to the sarcolemma (bold arrows) is expected to normalize substrate utilization and improve contractile function.
Subcellular protein trafficking is a complex process regulated by several proteins, including the family of vesicle-associated membrane proteins (VAMPs) which consists of 8 members. VAMP isoforms bring specificity to both organs and membrane proteins (CD36 vs. GLUT4). Thus, some VAMP isoforms are required for both CD36 and GLUT4 translocation, others are involved in only CD36 or GLUT4 trafficking36. These findings suggest the possibility of using VAMPs to manipulate specifically CD36-mediated fatty acid uptake without affecting GLUT4-mediated glucose uptake36.
Another protein involved in subcellular trafficking of substrate transporters is vacuolar H+-activated ATPase (v-ATPase), a multimeric protein complex present in the membrane of endosomes. By its proton pumping activity v-ATPase is responsible for the acidic lumen of endosomes37. Recent studies have disclosed that excess fatty acids inhibit v-ATPase activity causing the endosomes to lose their acidification, which in turn causes an increased translocation of CD36 from endosomes to the sarcolemma without affecting the sarcolemmal content of GLUT438. As a result, v-ATPase is part of a feed-forward cycle whereby lipid overexposure increases CD36-mediated fatty acid uptake, further impairing v-ATPase activity and increasing the translocation of CD36 to the sarcolemma12. Compounds able to re-activate v-ATPase could stop such feed-forward cycle and thus be used to lower sarcolemmal CD36 content and consequently the rate of fatty acid uptake.
Conclusions
Sarcolemmal substrate transporters, in particular, fatty acid transporter CD36 and glucose transporter GLUT4, are now recognized as representing the rate-governing kinetic step of myocardial substrate flux and, therefore, play pivotal roles in myocardial metabolism and energy provision. While mechanistic insight has been obtained mostly in studies with experimental animals and cell models, importantly, their overall roles also have been confirmed in patient studies13,39,40. Given their important roles, not surprisingly, evidence is accumulating that CD36 and GLUT4 also are involved in dysregulated cardiac metabolism associated with the pathogenesis of various cardiac diseases. The latter notion suggests the use of substrate transporters are target for metabolic modulation therapy. Particularly, manipulating the subcellular recycling of CD36 and of GLUT4 appears a promising approach because the involvement of tissue-specific and substrate-specific trafficking proteins in the recycling of CD36 and GLUT4 would allow the separate modulation of fatty acid and glucose uptake. Recent observations underscore the feasibility of this approach.
Alternative approaches to specifically alter either myocardial fatty acid or glucose uptake and utilization may also be worthwile to explore. For instance, PPARα agonists would upregulate specifically the expression of genes implicated in lipid utilization, including CD36, without affecting genes encoding proteins involved in glucose uptake and utilization12. Another potential approach is to exploit the emerging role of post-translational modifications of both CD36 and GLUT4 in their localization and functioning. Thus, CD36 undergoes a number of post-translational modifications including palmitoylation, O-GlcNAcylation and ubiquitination, which may influence its subcellular recycling and/or functioning41, while GLUT4 trafficking (in adipocytes) is regulated, among others, by (de)ubiquitination42. Disclosure of the putatively complex role of post-translational modifications on CD36 and GLUT4 functioning is likely to generate additional approaches for their application as metabolic modulation targets.
While long-chain fatty acids and glucose are the main substrates for cardiac energy provision, the heart is known to be a metabolic omnivore and will use virtually all types of substrates so as to assure an optimal cardiac performance43. Thus, the healthy heart continuously uses lactate and under certain (patho)physiological conditions also ketone bodies and amino acids. Interestingly, these substrates are also taken up into cardiomyocytes by substrate-specific transporters, i.e., monocarboxylate transporters (MCT) for lactate and ketone bodies, and L-type transporters and/or cationic transporters for amino acids34. Although knowledge on the involvement and mechanism of action of these other transporters is only beginning to be obtained, it would be of interest to explore whether these transporters could also be applied as targets for metabolic modulation therapy.
Acknowledgements
MN was supported by the Dutch Heart Foundation, Dekker grant # 2019T041.
Conflict of interest
No potential conflict of interest relevant to this article was reported.
References
- Boudina S, Abel ED. Diabetic cardiomyopathy, causes and effects. Reviews on Endocrinology and Metabolic Disorders. 2010; 11: 31-39.
- Wende AR, Brahma MK, McGinnis GR, et al. Metabolic origins of heart failure. JACC Basic and Translational Science. 2017; 2: 297-310.
- Chen L, Song J, Hu S. Metabolic remodeling of substrate utilization during heart failure progression. Heart Failure Review. 2019; 24: 143-154.
- Noordali H, Loudon BL, Frenneaux MP, et al. Cardiac metabolism – A promising therapeutic target for heart failure. Pharmacology and Therapeutics. 2018; 182: 95-114.
- Rosano G, Vitale C. Metabolic modulation of cardiac metabolism in heart failure. Cardiac Failure Review. 2018; 4: 99-103.
- Steggall A, Mordi IR, Lang CC. Targeting metabolic modulation and mitochondrial dysfunction in the treatment of heart failure. Diseases. 2017; 5: 14.
- Levelt EE, Gulsin G, Neubauer S, et al. Diabetic cardiomyopathy: pathophysiology and potential metabolic interventions state of the art review. European Journal of Endocrinology. 2018; 178: R127-R139.
- Birkenfeld A, Jordan J, Dworak M, et al. Myocardial metabolsm in heart failure: Purinergic signaling and other metabolic concepts. Pharmacology & Therapeutics. 2019; 194: 132-144.
- Holubarsch CJ, Rohrbach M, Karrasch M, et al. A double-blind randomized multicenter clinical trial to evaluate the efficacy and safety of two doses of etomoxir in comparison with placebo in patients with moderate congestive heart failure: the ERGO (etomoxir for the recovery of glucose oxidation) study. Clinical Science. 2007; 113: 205-212.
- Kerr M, Dodd MS, Heather LC. The ‘Goldilocks zone’ of fatty acid metabolism; to ensure that the relationship with cardiac function is just right. Clinical Science. 2017; 131: 2079-2094.
- Glatz JFC, Nabben M, Young ME, et al. Re-balancing cellular energy substrate metabolism to mend the failing heart. Biochimica et Biophysica Acta. 2020; in press. doi.or/10.1016/j.bbadis.2019.165579.
- Glatz JFC, Luiken JJFP. Dynamic role of the transmembrane glycoprotein CD36 (SR-B2) in cellular fatty acid uptake and utilization. Journal of Lipid Research. 2018; 59: 1084-1093.
- Klip A, McGraw TE, James DE. Thirty sweet years of GLUT4. Journal of Biological Chemistry. 2019; 294: 11369-11381.
- Bonen A, Luiken JJFP, Arumugam Y, et al. Acute regulation of fatty acid uptake involves the cellular redistribution of fatty acid translocase. Journal of Biological Chemistry. 2000; 275: 14501-14508.
- Habets DDJ, Coumans WA, Voshol PJ, et al. AMPK-mediated increase in myocardial long-chain fatty acid uptake critically depends on sarcolemmal CD36. Biochemical and Biophysical Research Communications 2007; 355: 204-210.
- Abumrad NA, Goldberg IJ. CD36 actions in the heart: Lipids, calcium, inflammation, repair and more? Biochimica et Biophysica Acta. 2016; 1861: 1442-1449.
- Saddik M, Gamble J, Witters LA, et al. Acetyl-CoA carboxylase regulation of fatty acid oxidation in the heart. Journal of Biological Chemistry. 1993; 268: 25836-25845.
- Luiken JJFP, Niessen HE, Coort SLM, et al. Etomoxir-induced partial carnitine palmitoyltransferase-I (CPT-I) inhibition in vivo does not alter cardiac long-chain fatty acid uptake and oxidation rates. Biochemical Journal 2009; 419: 447-455.
- He L, Kim T, Long Q, et al. Carnitine palmitoyltransferse-1b deficiency aggravates pressure overload-induced cardiac hypertrophy caused by lipotoxicity. Cicrulation. 2012; 126: 1705-1716.
- Glatz JFC, Nabben N, Heather LC, et al. Regulation of the subcellular trafficking of CD36, a major determinant of cardiac fatty acid utilization. Biochimica et Biophysica Acta. 2016; 1861: 1461-1471.
- Lundsgaard AM, Fritzen AM, Nicolaisen TS, et al. Glucometabolic consequences of acute and prolonged inhibition of fatty acid oxidation. Journal of Lipid Research. 2020; 61: 10-19.
- Abel ED. Glucose transport in the heart. Frontiers in Bioscience. 2004; 9: 201-215.
- Sala-Rabanal M, Hirayama BA, Ghezzi C, et al. Revisiting the physiological roles of SGLTs and GLUTs using positron emission tomography in mice. Journal of Physiology. 2016; 594: 4425-4438.
- Ouwens DM, Diamant M, Foddor M, et al. Cardiac contractile dysfunction in insulin-resistant rats fed a high fat diet is associated with elevated CD36-mediated fatty acid uptake and esterification. Diabetologia 2007; 50: 1938-1948.
- Luiken JJFP, Arumugam Y, Dyck DJ, et al. Increased rates of fatty acid uptake and plasmalemmal fatty acid transporters in obese Zucker rats. Journal of Biological Chemistry. 2001; 276: 40567-40573.
- Aguer C, Mercier J, Man CY, et al. Intramyocellular lipid accumulation is associated with permanent relocation ex vivo and in vitro of fatty acid translocase (FAT)/CD36 in obese patients. Diabetologia. 2010; 53: 1151-1163.
- Zhu B, Li MY, Lin Q, et al. Lipid oversupply induces CD36 sarcolemmal translocation via dual modulation of PKCζ and TBC1D1: an early event prior to insulin resistance. Theranostics. 2020; 10: 1332-1354.
- Bonen A, Jain SS, Snook LA, et al. Extremely rapid increase in fatty acid transport and intramyocellular lipid accumulation but markedly delayed insulin resistance after high fat feeding in rats. Diabetologia 2015; 58: 2381-2391.
- Liu Y, Neumann D, Glatz JFC, et al. Molecular mechanism of lipid-induced cardiac insulin resistance and contractile dysfunction. Prostaglandines, Leukotrienes and Essential Fatty Acids. 2018; 136: 131-141.
- Steinbusch LKM, Luiken JJFP, Vlasblom R, et al. Absence of fatty acid transporter CD36 protects against Western-type diet-related cardiac dysfunction following pressure overload in mice. American Journal of Physiology Endocrinology and Metabolism 2011; 301: E618-E628.
- Sung MM, Koonen DP, Soltys CL, et al. Increased CD36 expression in middle-aged mice contributes to obesity-related cardiac hypertrophy in the absence of cardiac dysfunction. Journal of Molecular Medicine. 2011; 89: 459-469.
- Yang J, Sambandam N, Han X, et al. CD36 deficiency rescues lipotoxic cardiomyopathy. Circulation Research. 2007; 100: 1208-1217.
- Angin Y, Steinbusch LKM, Simons PJ, et al. CD36 inhibition prevents lipid accumulation and contractile dysfunction in rat cardiomyocytes. Biochemecial Journal. 2012; 448: 43-53.
- Geraets IME, Glatz JFC, Luiken JJFP, et al. Pivotal role of membrane substrate transporters on the metabolic alterations in the pressure-overloaded heart. Cardiovascular Research. 2019; 115: 1000-1012.
- Dirkx E, van Eys GJ, Schwenk RW, et al. Protein kinase-D1 overexpression prevents lipid-induced cardiac insulin resistance. Journal of Molecular and Cellular Cardiology. 2014; 76:208-217.
- Schwenk RW, Dirkx E, Coumans WA, et al. Requirement for distinct vesicle-associated membrane proteins in insulin- and AMP-activated protein kinase (AMPK)-induced translocation of GLUT4 and CD36 in cultured cardiomyocytes. Diabetologia. 2010; 53: 2209-2219.
- McGuire C, Stransky L, Cotter K, et al. Regulation of V-ATPase activity. Frontiers in Bioscience. 2017; 22: 609-622.
- Liu Y, Steinbusch LKM, Nabben N, et al. Palmitate-induced vacuolar-type H+-ATPase inhibition feeds forward into insulin resistance and contractile dysfunction. Diabetes. 2017; 66: 1521-1534.
- Tanaka T, Nakata T, Oka T, et al. Defect in human myocardial long-chain fatty acid uptake is caused by FAT/CD36 mutations. Journal of Lipid Research. 2001; 42: 751-759.
- Melis M, Carta G, Pintus S, et al. Polymorphism re1761667 in the CD36 gene is associated to changes in fatty acid metabolism and circulating endocannabinoid levels distinctively in normal weigth and obese subjects. Frontiers in Physiology. 2017; 8: 1006.
- Luiken JJFP, Chanda D, Nabben M, et al. Post-translational modifications of CD36 (SR-B2): Implications for regulation of myocellular fatty acid uptake. Biochimica et Biophysica Acta. 2016; 1862: 2253-2258.
- Sadler JBA, Lamb CA, Welburn CR, et al. The deubiquinating enzyme USP25 binds tankyrase and regulates trafficking of the facilitative glucose transporter GLUT4 in adipocytes. Scientific Reports. 2019; 9: 4710.
- Stanley WC, Recchia FA, Lopaschuk GD. Myocardial substrate metabolism in the normal and failing heart. Physiological Reviews. 2005; 85: 1093-1129.