
Pathogenesis Of Portal Vein Thrombosis In Liver Cirrhosis: The Role of the ADAMTS13/VWF Unbalance
Monica Sacco1*, Stefano Lancellotti1*, Maria Basso1, Raimondo De Cristofaro1,2*
1Servizio Malattie Emorragiche e Trombotiche, Fondazione Policlinico Universitario “A. Gemelli”, IRCCS, Italy
2Istituto di Medicina Interna e Geriatria, Università Cattolica S. Cuore, Roma, Italy
Abstract
Increasing evidence shows a potential role of ADAMTS13 deficiency as a risk factor for the high prevalence of portal vein thrombosis (PVT) in cirrhotic patients. This deficiency, due to myofibroblastic transformation of hepatic stellate cells (HSCs), the source of ADAMTS13, is responsible for the prevalence of ultra large molecular weight multimers of von Willebrand factor (UL-VWF) in the hepatic microcirculation. This phenomenon would favor the pro-haemostatic function of VWF, which, together with an elevation of coagulation FVIII, which is associated to VWF, could sustain microcirculatory thrombosis in the liver. These phenomena, triggering an increase of the intra-hepatic pressure, would cause a slowdown of the portal flow, favoring the occurrence of PVT. Although this scenario is justified by retrospective observational clinical studies, it will be mandatory to clarify the ADAMTS13 expression in HSCs associated with the activity of plasma ADAMTS13 in different stage of liver diseases. Hence, a prospective clinical trial (ClinicalTrials.gov Identifier: NCT03322696) is ongoing to unravel the linkage between all the actors involved in the complex phenomenon of PVT occurring in cirrhosis.
Introduction
The liver plays a central role in the control of blood coagulation, as it produces the pro-coagulant vitamin K dependent factors II, VII, IX, X, in addition to factor FV, FXIII and fibrinogen, as well as the physiological anticoagulants protein C, protein S, protein Z and antithrombin1. Moreover, the hepatic reticuloendothelial system contributes to regulation of coagulation by clearing activated clotting and fibrinolytic products, most notably tissue plasminogen activator (tPA), whose concentration is often elevated in acute and chronic liver diseases1. In the context of this scenario, the richness of endothelial cells pertaining to liver circulation aids to further regulate haemostasis by producing and secreting von Willebrand factor (VWF) multimers, tPA, thrombomodulin, tissue factor pathway inhibitor (TFPI) and urokinase1. Thus, it is not surprising that numerous coagulation abnormalities were reported in clinical settings characterized by acute and chronic liver disorders. Viewpoints concerning the clinical consequences of the hemostatic disorders in patients with liver failure have consistently changed over the last decade. Patients with chronic liver failure and abnormal routine coagulation tests, such as prothrombin time, do not necessarily undergo an increased bleeding tendency. On the contrary, thrombotic complications may even occur in these patients1,2. This apparently unexpected phenomenon may be even responsible for the progression of liver failure due to intrahepatic thrombosis3,4. Among the relevant clinical complications deriving from coagulation abnormalities occurring during chronic liver disorders, thrombosis of the portal vein (PVT) is one of the most severe complications. In fact, this vascular disorder complicating liver cirrhosis, causes a deterioration of the general clinical conditions of the patient and may often hamper the possibility to receive an orthotopic liver transplantation (OLT). The prevalence of PVT in cirrhosis is associated with worsening of liver function and has been reported to be as high as 15 % at pathological analysis of the explanted liver in patients who undergo OLT5. Several studies have investigated the association of classic thrombophilia risk factors with occurrence of PVT in cirrhosis and chronic liver diseases6-11. Unfortunately, no clear association was found in this field. The reduction of blood flow in the portal vein was the only factor to be significantly associated with the development of PVT12,13. The possible contribution of VWF and ADAMTS13 to the pathogenesis of PVT in this setting was addresses in a few studies only (see below). The scientific rationale for investigating the role of ADAMTS13/VWF in the pathogenesis of PVT arises from the fact that ADAMTS13 is produced and secreted mostly by the stellate cells of the liver14. The fibroblastic differentiation of the stellate cells causes a almost complete defect of ADAMTS13 production. An relevant functional consequence on VWF activity stems from this phenomenon. In fact, the functional capacity of VWF is normally strictly regulated in the circulation by the zinc-protease ADAMTS13, as VWF reactivity towards platelets is directly proportional to its multimeric size15. The cleavage by ADAMTS13 of the peptide bond between Tyr1605 and Met1606 in the A2 domain of VWF, reduces in fact the molecular weight of the multimer and limits its pro-haemostatic potential. The relevance of this regulation is so high that patients with congenital or acquired ADAMTS13 deficiency undergo severe thrombotic episodes in the microvasculature, causing the disorders globally referred to as thrombotic microangiopathies (TMAs)16,17. TMAs are characterized by microangiopathic hemolytic anemia with red cell fragmentation, thrombocytopenia and signs of organ dysfunction due to disturbed microcirculation. The severe forms of thrombotic microangiopathies referred to as Thrombotic Thrombocytopenic Purpura (TTP) may be congenital, due to mutations of the ADAMTS13 gene (named also Upshaw-Schulman syndrome)18 or acquired, when associated with the production of anti-ADAMTS13 autoantibodies16. In both cases, the lack of ADAMTS13 leads to the persistence of ultralarge (UL) VWF multimeres. In the presence of additional triggers causing shear stress and unfolding of VWF (pregnancy, infections, certain drugs, surgery, etc.) enhanced platelet aggregation with the UL-VWF multimers occurs. These platelet aggregates affect the blood flow in the microcirculation and cause organ damage and clinical symptoms19.
The Role of Flow
The classic thrombophilia risk factors were investigated as main cause of PVT occurrence, but a really strong association was never found6,9,10, although the G20210A mutation of prothrombin gene was proposed as a risk factor for PVT in cirrhosis in a single study10. Our center also addressed this issue in a previous study and, although an extensive research was carried out on a wide spectrum of thrombophilic risk factors potentially associated with PVT, only a low blood flow (<15 cm/sec) in portal vein was found significantly associated with PVT in a consecutive series of cirrhotic patients12. Low blood flow belongs to the classic Virchow’s triad of factors strongly associated with venous thrombosis and this finding suggests that secondary haemostasis is strongly engaged in the pathogenesis of PVT, as low blood flow facilitates the local persistence of high concentrations of activated coagulation factors. Although this finding provided a significant association between portal vein flow and PVT, no mechanistic explanation for the thrombotic complication emerged from that study. In particular, whether the reduced blood flow in the portal vein is either the cause or the effect of PVT was not explained in that study, due to its merely observational nature.
3The Role of ADAMTS13
Previous studies showed that the principal site of ADAMTS13 production is liver and, in particular, the liver stellate cells (HSCs)14. HSCs are in the space of Disse adjacent to endothelial cells of hepatic sinusoids. This histological location is of relevance because HSCs, through production of ADAMTS13, can regulate the sinusoidal blood flow and maintain homeostasis in the microenvironment of the hepatic sinusoid20. Moreover, HSCs have other functions, including vitamin A storage and liver fibrogenesis20. ADAMTS13, through proteolytic processing of VWF, synthesized and secreted by endothelial cells of the liver sinusoids, limits the pro-hemostatic and procoagulant activity of VWF. In fact, the higher the molecular weight of the VWF multimers, the higher their ability to interact with platelets and sub-endothelial collagen. Previous studies, including one of our research group, showed that liver cirrhosis is associated with a significant decrease of ADAMTS13 plasma levels, especially in advanced stages of the disease21-29. Thus, the transformation of the HSCs in cirrhotic liver is really associated with a decreased production of ADAMTS13. Whether this association is sufficient per se to promote PVT or it acts through a perturbed homeostasis of VWF is still to be unraveled.
The Role of VWF and FVIII
In patients with liver cirrhosis, a remarkably high level of plasma VWF has been reported by different research groups30,31. Immuno-histochemical studies showed that VWF multimers are present in the sinusoidal lining cells and at the scar-parenchyma interface in cases of liver cirrhosis32. The presence of VWF is particularly abundant in the sinusoids at early stages of alcoholic steatohepatitis33. This finding reflects the capillarization of the sinusoidal endothelial cells, a phenomenon likely deriving also from the activity of VWF multimers on vasculature34. On the other hand, chronic liver diseases leading to cirrhosis are sustained by inflammatory mechanisms that are associated to massive endothelial activation, secretion of VWF, and adhesion and deposition of VWF-platelet thrombi in the vasculature. This mechanism can involve not only small vessels, but large arterial as well as venous vessels35. Considering the global scenario whereby a strong decrease of ADAMTS13 production parallels a significant increase of UL-VWF multimers synthesized by transformed vascular endothelial cells, the deficiency of plasma ADAMTS13 activity due to transformed HSCs in cirrhotic liver may play an important role in sinusoidal microcirculatory disturbances. Thus, this pathological phenomenon would represent a risk factor for both microcirculatory disturbances and thrombosis as well as subsequent development of liver injury. The microcirculatory disturbances may also influence the portal vein flow, which could be slowed down by the thrombotic phenomenon generated in the liver microcirculation, as showed in an observational study12,13. Moreover, another aspect remains to be clarified to explain the potential role of VWF in the pathogenesis of PVT and concerns the role of coagulation FVIII. This issue stems from the strong interaction between VWF and FVIII36. The striking increase of VWF levels in cirrhotic patients is accompanied in fact by FVIII elevation37. It is known that a persistent elevation of FVIII is now considered a risk factor for venous thromboembolism and even in cirrhosis37,38. FVIII may compensate the decrease of vitamin K-dependent factors level in cirrhosis, accelerating the coagulation pathway in such condition37. The contemporary decrease of ADAMTS13 activity, which favors the presence of ultra-large VWF multimers, the strong increase of FVIII level in the presence of low portal vein flow may thus represent a risk factor for the development of PVT in cirrhotic patients. The flowchart shown in Fig. 1 depicts this pathogenetic mechanism.
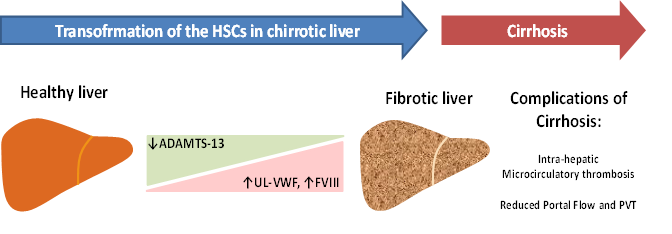
Figure 1: Flowchart of the proposed pathogenetic mechanisms leading to portal vein thrombosis in liver cirrhosis. Abbreviations: HSCs: hepatic stellate cells; UL-VWF: ultra-large von Willebrand factor; FVIII: coagulation factor VIII.
Conclusions
Although there is quite compelling evidence that outlines a potential role of ADAMTS13 deficiency for PVT occurrence in cirrhosis, it will be mandatory to clarify the intralobular heterogeneity of ADAMTS13 expression in HSCs associated with the activity of plasma ADAMTS13 in different stage of liver diseases. Hence, prospective clinical trials are needed to unravel the linkage between all the actors involved in a complex phenomenon, such as PVT in cirrhosis. To address this point, our research group is performing a prospective clinical trial (ClinicalTrials.gov Identifier: NCT03322696), aimed at assessing the mechanistic roles of ADAMTS13, VWF levels, thrombophilia risk factors, and portal blood flow in the pathogenesis of PVT in a large cohort of cirrhotic patients.
Acknowledgments
Financial support from the Catholic University School of Medicine (Linea D1 -2017) and from Baxalta/Shire are gratefully acknowledged.
References
- Tripodi A, Mannucci PM. The coagulopathy of chronic liver disease. N Engl J Med. 2011; 365: 147-56.
- Lisman T, Porte RJ. Rebalanced hemostasis in patients with liver disease: evidence and clinical consequences. Blood. 2010; 116: 878-85.
- Anstee QM, Goldin RD, Wright M, et al. Coagulation status modulates murine hepatic fibrogenesis: implications for the development of novel therapies. J Thromb Haemost. 2008; 6: 1336-43.
- Villa E, Camma C, Marietta M, et al. Enoxaparin prevents portal vein thrombosis and liver decompensation in patients with advanced cirrhosis. Gastroenterology. 2012; 143: 1253-60 e4.
- Francoz C, Belghiti J, Vilgrain V, et al. Splanchnic vein thrombosis in candidates for liver transplantation: usefulness of screening and anticoagulation. Gut. 2005; 54: 691-7.
- Janssen HL, Meinardi JR, Vleggaar FP, et al. Factor V Leiden mutation, prothrombin gene mutation, and deficiencies in coagulation inhibitors associated with Budd-Chiari syndrome and portal vein thrombosis: results of a case-control study. Blood. 2000; 96: 2364-8.
- Primignani M, Martinelli I, Bucciarelli P, et al. Risk factors for thrombophilia in extrahepatic portal vein obstruction. Hepatology. 2005; 41: 603-8.
- Parikh S, Shah R, Kapoor P. Portal vein thrombosis. Am J Med. 2010; 123: 111-9.
- D'Amico M, Pasta F, Pasta L. Thrombophilic genetic factors PAI-1 4G-4G and MTHFR 677TT as risk factors of alcohol, cryptogenic liver cirrhosis and portal vein thrombosis, in a Caucasian population. Gene. 2015; 568: 85-8.
- Amitrano L, Guardascione MA, Ames PR, et al. Increased plasma prothrombin concentration in cirrhotic patients with portal vein thrombosis and prothrombin G20210A mutation. Thromb Haemost. 2006; 95: 221-3.
- Zoller B, Li X, Sundquist J, et al. Familial risks of unusual forms of venous thrombosis: a nationwide epidemiological study in Sweden. J Intern Med. 2010; 270: 158-65.
- Zocco MA, Di Stasio E, De Cristofaro R, et al. Thrombotic risk factors in patients with liver cirrhosis: correlation with MELD scoring system and portal vein thrombosis development. J Hepatol. 2009; 51: 682-9.
- Ponziani FR, Zocco MA, Campanale C, et al. Portal vein thrombosis: insight into physiopathology, diagnosis, and treatment. World J Gastroenterol. 2010; 16: 143-55.
- Uemura M, Tatsumi K, Matsumoto M, et al. Localization of ADAMTS13 to the stellate cells of human liver. Blood. 2005; 106: 922-4.
- Fujikawa K, Suzuki H, McMullen B, et al. Purification of human von Willebrand factor-cleaving protease and its identification as a new member of the metalloproteinase family. Blood. 2001; 98: 1662-6.
- George JN. Clinical practice. Thrombotic thrombocytopenic purpura. N Engl J Med. 2006; 354: 1927-35.
- Hrdinova J, D'Angelo S, Graca NAG, et al. Dissecting the pathophysiology of immune thrombotic thrombocytopenic purpura: interplay between genes and environmental triggers. Haematologica. 2018; 103: 1099-109.
- Hommais A, Rayes J, Houllier A, et al. Molecular characterization of four ADAMTS13 mutations responsible for congenital thrombotic thrombocytopenic purpura (Upshaw-Schulman syndrome). Thromb Haemost. 2007; 98: 593-9.
- George JN, Nester CM. Syndromes of thrombotic microangiopathy. N Engl J Med. 2014; 371: 654-66.
- Geerts A. History, heterogeneity, developmental biology, and functions of quiescent hepatic stellate cells. Semin Liver Dis. 2001; 21: 311-35.
- Uemura M, Fujimura Y, Ko S, et al. Determination of ADAMTS13 and Its Clinical Significance for ADAMTS13 Supplementation Therapy to Improve the Survival of Patients with Decompensated Liver Cirrhosis. Int J Hepatol. 2011; 2011: 759047.
- Takaya H, Uemura M, Fujimura Y, et al. ADAMTS13 activity may predict the cumulative survival of patients with liver cirrhosis in comparison with the Child-Turcotte-Pugh score and the Model for End-Stage Liver Disease score. Hepatol Res. 2012; 42: 459-72.
- Hamed AA, Darwish YW, El-Sayed MH. ADAMTS13 Levels in Young Patients With beta-Thalassemia Major: Relation to Hepatitis C Virus Infection, Liver Cirrhosis, and Iron Overload. Clin Appl Thromb Hemost. 2015; 21: 527-32.
- Reuken PA, Kussmann A, Kiehntopf M, et al. Imbalance of von Willebrand factor and its cleaving protease ADAMTS13 during systemic inflammation superimposed on advanced cirrhosis. Liver Int. 2015; 35: 37-45.
- Uemura M, Fujimura Y, Ko S, et al. Pivotal role of ADAMTS13 function in liver diseases. Int J Hematol. 2010; 91: 20-9.
- Uemura M, Fujimura Y, Matsumoto M, et al. Comprehensive analysis of ADAMTS13 in patients with liver cirrhosis. Thromb Haemost. 2008; 99: 1019-29.
- George JN. Clinical insights from observations on ADAMTS13 deficiency in liver cirrhosis. Thromb Haemost. 2008; 99: 987-8.
- Yagita M, Uemura M, Nakamura T, et al. Development of ADAMTS13 inhibitor in a patient with hepatitis C virus-related liver cirrhosis causes thrombotic thrombocytopenic purpura. J Hepatol. 2005; 42: 420-1.
- Lancellotti S, Basso M, Veca V, et al. Presence of portal vein thrombosis in liver cirrhosis is strongly associated with low levels of ADAMTS-13: a pilot study. Intern Emerg Med. 2016; 11: 959-67.
- La Mura V, Reverter JC, Flores-Arroyo A, et al. Von Willebrand factor levels predict clinical outcome in patients with cirrhosis and portal hypertension. Gut. 2011; 60: 1133-8.
- Mannucci PM. von Willebrand factor: a marker of endothelial damage. Arterioscler Thromb Vasc Biol. 1998; 18: 1359-62.
- Knittel T, Neubauer K, Armbrust T, et al. Expression of von Willebrand factor in normal and diseased rat livers and in cultivated liver cells. Hepatology. 1995; 21: 470-6.
- Urashima S, Tsutsumi M, Nakase K, et al. Studies on capillarization of the hepatic sinusoids in alcoholic liver disease. Alcohol Alcohol Suppl. 1993; 1B: 77-84.
- Randi AM, Smith KE, Castaman G. von Willebrand factor regulation of blood vessel formation. Blood. 2018; 132: 132-40.
- Chen J, Chung DW. Inflammation, von Willebrand factor, and ADAMTS13. Blood. 2018; 132: 141-7.
- Dimitrov JD, Christophe OD, Kang J, et al. Thermodynamic analysis of the interaction of factor VIII with von Willebrand factor. Biochemistry. 2012; 51: 4108-16.
- Sinegre T, Duron C, Lecompte T, et al. Increased factor VIII plays a significant role in plasma hypercoagulability phenotype of patients with cirrhosis. J Thromb Haemost. 2018; 16: 1132-40.
- Golder M, Mewburn J, Lillicrap D. In vitro and in vivo evaluation of the effect of elevated factor VIII on the thrombogenic process. Thromb Haemost. 2013; 109: 53-60.