
Protective Mechanism of Thioredoxin-1 against Atherosclerotic Endothelial Injury Induced by Ox-LDL
Lu Feng1, Gexin Zhao2*, Beidong Chen1*
*1The MOH Key Laboratory of Geriatrics, Beijing Hospital, National Center of Gerontology, Beijing, 100730, China
*2Technology Center for Genomics & Bioinformatics. Department of Pathology & Laboratory Medicine, David Geffen School of Medicine, University of California, California 90095, USA
Abstract
Atherosclerosis is a chronic inflammation disease that is initiated by endothelial cell injury. Hyperlipidemia is an independent risk factor for atherosclerosis. Oxidized low-density lipoprotein (ox-LDL) is a major component of hyperlipidemia and contributes to atherosclerosis. Ox-LDL and ox-LDL-related reactive oxygen species (ROS) and inflammation have deleterious effects on vascular endothelial cells. Previous studies have demonstrated that thioredoxin-1 (Trx) is one of the key regulators of intracellular redox, which may play a role in the pathogenesis of atherosclerosis. This review will summarize the protective mechanisms of Trx on endothelial injury induced by ox-LDL.
Introduction
Atherosclerosis is the main cause of coronary heart disease, cerebral infarction, peripheral vascular diseases. It is a chronic inflammation disease that is initiated by endothelial cell injury. Lipid metabolic disorders are the basis of atherosclerotic lesions. Abundant data link hypercholesterolaemia to atherogenesis. However, recently we have demonstrated that inflammatory mechanisms couple dyslipidaemia to atheroma formation. Over the past decade, we have come to appreciate a prominent role for inflammation in atherosclerosis and its complications, and put forward the notion that atherosclerosis is a chronic inflammation. In early atherogenesis, pro-inflammatory chemokines recruit leukocytes and induce expression of pro-inflammatory cytokines which contribute greatly to atherogenesis1.
Among the factors contributing to atherosclerosis, reactive oxygen species (ROS) is a pivot to regulate many signal pathways. ROS are normal products of cellular redox metabolism. It is believed that ROS serve as subcellular messengers in gene regulatory and signal transduction pathways. Whereas when ROS are generated more than scavenging, they accumulated. Once the cellular redox homeostasis is broken out, oxidative stress occurred. ROS can regulate some inflammatory gene’s expression via signal transduction pathways in addition to direct damnification of cells, and lead to a series of pathological and physiological courses. Endothelial dysfunction and activation are crucial factors to initiate atherosclerosis which can be contributed by various factors such as ox-LDL, advanced glycation end-products (AGEs), and Angiotensin-2 etc2. Ox-LDL disrupts the antioxidant and secretory activities of vascular endothelium3, and stimulates vascular O2- formation presumably via activation of the membrane-bound nicotinamide adenine dinucleotide phosphate (NADPH) oxidase4, thus being considered as an important risk factor for endothelial injury. A large body of evidence suggests that increase of ROS is involved in the genesis of both endothelial vasodilator dysfunction and endothelial activation5. Of the several potential sources of ROS in vasculature, the endothelial NADPH oxidase family is a major contributor to ROS in atherosclerosis.
ROS level is tightly controlled in vasculature. Thioredoxin (Trx) is small-molecule protein, which exists widely in eukaryotic and prokaryotic organisms. Trx, thioredoxin reductase (TrxR) and NADPH comprise the thioredoxin system, which is one of the most important thiol oxidoreductase systems. Human thioredoxin is a 12-kDa protein with redox-active dithiol, its active site -CysGly-Pro-Cys- contains two cysteines (dithiol) undergoing reversible redox change between an oxidized disulfides (-S-S-) and a reduced dithiol (-SH, -SH)6. Currently, three isoforms of human Trx have been identified. Trx1 is a 104-amino acid protein that is typically found in the cytoplasm but has also been identified in the nucleus of normal endometrial stromal cells, tumor cells, and primary solid tumors7. Trx2 usually exists in mitochondria, it is a 166-amino acid protein that contains a 60-amino acid NH2-terminal translocation sequence8. The third isoform SpTrx is highly expressed in testicular tissue, which only exists in the Golgi apparatus in mammalian spermatocytes and sperm cells9. Unless otherwise indicated, Trx refers to Trx1 in this review. Evidence indicates that Trx may play a role in the pathogenesis of atherosclerosis10,11,12,13,14, including the control of cellular redox balance, and the therapeutic approaches targeting the Trx system are emerging.
This review will explore the protective mechanisms of Trx on endothelial injury induced by ox-LDL, and may provide new clues for atherosclerosis prevention and therapy.
Trx reduces the activity of NADPH oxidase
ox-LDL is well known to play a crucial role in the initiation and progression of atherosclerosis, which can be considered an inflammatory disease15. Under the stimulation of ox-LDL, vascular endothelial cells can increase the generation of ROS and the expression of inflammatory proteins10,11. ROS-induced oxidative stress is closely related to endothelial dysfunction in humans and rats5,16,17. Of the several potential sources of ROS in the vasculature, the endothelial NADPH oxidase family is a major contributor to ROS associated with atherosclerosis18. Nox4 and Nox2 are functional NADPH oxidases isoforms, expressed in endothelial cells. They are the major sources of ROS in endothelial cells and superabundant ROS are implicated in both vasodilator dysfunction and the modulation of redox-sensitive signaling pathways19. P22phox, a membrane associated component of NADPH oxidase, co-localizes with and directly binds to Nox4. The protein level and stability of p22phox have been reported to be necessary for Nox4 activity20. Nox4 does not require the binding of cytosolic proteins (p47phox, p67phox, and Rac1) for ROS production and instead produces ROS constitutively21. Thus, p22phox may be an important regulator of Nox4. Nox2 is another isoform of NADPH oxidase, which also exists in endothelial cells. Nox2 is a highly glycosylated protein, and its activation requires interactions with other membrane (p22phox) and cytosolic (p47phox, p67phox, p40phox, and Rac1/2) components. Rac1 is a cytosolic component of NADPH oxidase, which can bind to Nox2 and affect Nox2 activity. A previous study indicated that Trx levels are associated with NADPH oxidase activity in vivo and in vitro13. Recently, our lab reported that Trx decreased NADPH oxidase activity in ox-LDL-stimulated human umbilical vein endothelial cells (HUVECs) by detecting NADPH consumption in the NADPH (100 mM) incubated homogenate of supernatant yielded the plasma membrane-enriched fractions. Genetic gain-of-function and loss-of-function studies showed that Trx suppressed ox-LDL induced Nox4 and p22phox expression. A co-immunoprecipitation assay indicated that Trx1 decreased Nox4-p22phox complex level during ox-LDL stimulation. Transient transfection of Nox4 and p22phox significantly increased intracellular ROS generation, which could be blocked by Trx overexpression. In addition, Trx overexpression also prevented ox-LDL-induced Nox2 and Rac1 protein levels (Fig. 1).
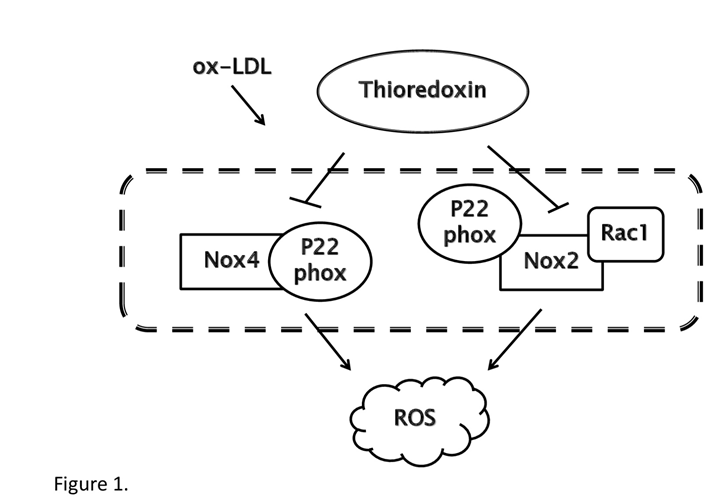
Figure 1: Schematic model of the signaling pathway of Trx. Trx inhibits the generation of ROS induced by ox-LDL. Ox-LDL activates NADPH oxidase, which acts as the main source of ROS in endothelial cells and induces signaling cascade that results in inflammation and endothelial cells dysfunction. In cytoplasm of endothelial cells, Trx decreases Nox4 and p22phox level via physically interaction with the Nox4-p22phox complex. Trx also suppresses Nox2 and Rac1 level stimulated by ox-LDL. Thus Trx down-regulates amount of ROS and protects endothelial cells.
Our data indicated that Trx inhibited NADPH activity via suppressing Nox4, ph22phox, Nox2, Rac1 expression, and decreasing the level of Nox4-p22phox complex in ox-LDL treated HUVCEs and may prevent the initiation of atherosclerosis by attenuating exceeding ROS production12 .
Trx regulates AP-1 and Smad3 pathways
Trx regulates a number of redox-sensitive cellular signal transduction molecules and transcription factors including nuclear factor-kappa B (NF-κB), activator protein-1 (AP-1), tumor protein 53 (p53), PEBP222, Smad311 and others. Redox Factor 1 (Ref-1) is a 36.5 kDa protein known as apurinic/apyrimidinic endonuclease (APE1) and was first described as an important enzyme in the base excision repair (BER) pathway. Moreover, Ref-1 has a separate N-terminal domain with redox activity23. Its activity has been shown to enhance the DNA binding ability of transcription factors such as NF-kB24,25, p5326, hypoxia inducible factor-1α (HIF-1α)27 and AP-128. We focused on AP-1 pathways. Trx has been shown to bind to Ref-1 in vitro and through mammalian two–hybrid assays in Cos-7 cells29. Other studies have further implicated that Trx can cooperate with Ref-1 within the nucleus of mammalian cells to enhance AP-1 activity22,30. Our lab reported that overexpression of Trx in ox-LDL stimulated endothelial cells inhibited c-Jun translocation into the nucleus, knockdown of Trx extremely enhanced nuclear Ref-1 expression and increased the DNA binding activity of AP-1. Luciferase assay also certified that Trx inhibited AP-1 activity in ox-LDL treated endothelial cells10. The above experimental results seem to be inconsistent, but this may be due to different cell types and stimulus conditions. The expression of Trx in human vascular endothelial cells affects the nuclear translocation of Ref-1 and AP-1 subunit c-Jun, thereby inhibiting the activity of AP-1.
Smad transcription factors are specific downstream mediators of the transforming growth factor β (TGF-β) signaling pathway. Smad3 belongs to receptor-regulated Smads (R-Smads) and can be activated by TGF-β and activin receptors. In the process of atherosclerotic plaque formation, TGF-β/Smad3 signaling pathway plays a role of anti-inflammatory protection. TGF-β prevents the ox-LDL-induced expression of adhesion molecules31 and contributes to plaque stabilization32. Additionally, in vascular cells, cholesterol suppressed TGF-β signaling by increasing lipid rafts and the caveolae accumulation of TGF-β receptors33. Although the TGF-β/Smad pathway has been shown to have protective, anti-inflammatory effects on cells that are important to atherosclerotic lesion formation [8], it remains unclear how Smad3 contributes to ox-LDL stimulation in HUVECs. Recently, we found that overexpression of functional Trx significantly inhibited VCAM-1 and ICAM-1 expression in ox-LDL stimulated HUVECs, which can be abolished by SIS3, a specific inhibitor of Smad3 phosphorylation. Further detected, we found that Trx enhanced Smad3 phosphorylation and nuclear translocation. A co-immunoprecipitation assay indicated that Smad3 continued to interact with Trx. This interaction might be responsible for the further nuclear translocation of active Smad3 (phosphorylated Smad3, pSmad3) in Trx-overexpressing HUVECs and activation of the Smad3 signaling pathway which presents a crucial role in atherogenesis11 (Fig. 2).
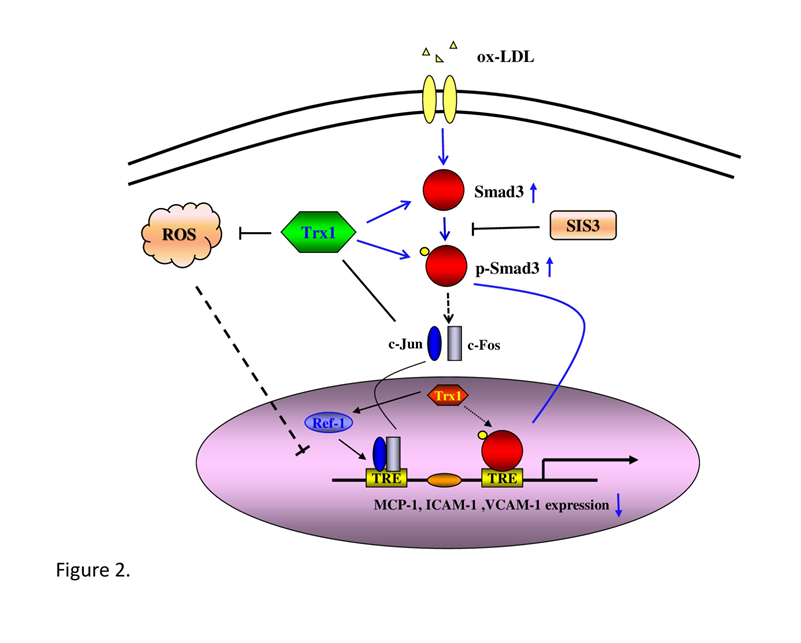
Figure 2: Mechanisms for Trx in the prevention of ox-LDL-induced endothelial cells dysfunction. Trx inhibits ox-LDL-induced adhesion and chemoattractant proteins expression via reducing ROS generation, c-Jun translocation and increasing Smad3 phosphorylation and translocation.
Trx inhibits the expression of inflammatory proteins
Atherosclerosis is a hypercholesterolemia-based and oxidative stress-associated chronic inflammatory disease. Endothelial cells express vascular cell adhesion molecule-1 (VCAM-1) and intercellular adhesion molecule-1 (ICAM-1) in response to cholesterol feeding selectively in areas prone to lesion formation [5, 6], which seem to play a major role in recruiting monocyte to the atheroma. Monocyte chemoattractant protein-1 (MCP-1) is a key mediator of monocyte infiltration and plays a crucial role in the pathogenesis of atherosclerosis. These inflammatory factors link the occurrence of hyperlipidemia and atherosclerosis, which are important initiators of atherosclerosis. Our study indicated that Trx could suppress VCAM-1, ICAM-1 expression and MCP-1 secretion in ox-LDL stimulated HUVECs10,11. Therefore, Trx may play an essential role of atheroprotection (Fig. 2).
Summary
In conclusion, our series of studies put forward new mechanisms of Trx for protecting endothelial cells against ox-LDL induced atherosclerosis, which may provide new clues for atherosclerosis prevention and treatment at the level of endothelial cells.
References
- Libby P. Inflammation in atherosclerosis. Nature. 2002; 420: 868-874.
- Mannarino E, Pirro M. Endothelial injury and repair: a novel theory for atherosclerosis. Angiology. 2008; 59: 69S-72S.
- Lubrano V, Balzan S. LOX-1 and ROS, inseparable factors in the process of endothelial damage. Free Radic Res. 2014; 48: 841-848.
- Galle j, Schneider R, Heinloth A, et al. Lp(a) and LDL induce apoptosis in human endothelial cells and in rabbit aorta: role of oxidative stress. Kidney Int. 1999; 55: 1450-1461.
- Higashi Y, Maruhashi T, Noma K, et al. Oxidative stress and endothelial dysfunction: clinical evidence and therapeutic implications. Trends Cardiovasc Med. 2014; 24: 165-169.
- Yodoi J, Matsuo Y, Tian H, et al. Anti-Inflammatory Thioredoxin Family Proteins for Medicare, Healthcare and Aging Care. Nutrients. 2017; 9.
- Powis G, Montfort WR. Properties and biological activities of thioredoxins. Annu Rev Biophys Biomol Struct. 2001; 30: 421-455.
- Zhang R, Al-Lamki R, Bai L, et al. Thioredoxin-2 inhibits mitochondria-located ASK1-mediated apoptosis in a JNK-independent manner. Circ Res. 2004; 94: 1483-1491.
- Miranda-Vizuete A, Ljung J, Damdimopoulos AE, et al. Characterization of Sptrx, a novel member of the thioredoxin family specifically expressed in human spermatozoa. J Biol Chem 276 (2001) 31567-31574.
- Chen B, Guan D, Cui ZJ, et al. Thioredoxin 1 downregulates MCP-1 secretion and expression in human endothelial cells by suppressing nuclear translocation of activator protein 1 and redox factor-1. Am J Physiol Cell Physiol. 2010; 298: C1170-1179.
- Chen B, Wang W, Shen T, et al. Thioredoxin1 downregulates oxidized low-density lipoprotein-induced adhesion molecule expression via Smad3 protein. PLoS One. 2013; 8: e76226.
- Chen B, Meng L, Shen T, et al. Thioredoxin attenuates oxidized low-density lipoprotein induced oxidative stress in human umbilical vein endothelial cells by reducing NADPH oxidase activity. Biochem Biophys Res Commun. 2017; 490: 1326-1333.
- Madrigal-Matute J, Fernandez-Garcia CE, Blanco-Colio LM, et al. Thioredoxin-1/peroxiredoxin-1 as sensors of oxidative stress mediated by NADPH oxidase activity in atherosclerosis. Free Radic Biol Med. 2015; 86: 352-361.
- Bechtel TJ, Weerapana E. From structure to redox: The diverse functional roles of disulfides and implications in disease. Proteomics. 2017; 17.
- Ross R. Atherosclerosis--an inflammatory disease. N Engl J Med. 1999; 340: 115-126.
- Ishikawa T, Seki K. The association between oxidative stress and endothelial dysfunction in early childhood patients with Kawasaki disease. BMC Cardiovasc Disord. 2018; 18: 30.
- Chang HH, Chien CY, Chen KH, et al. Catechins Blunt the Effects of oxLDL and its Primary Metabolite Phosphatidylcholine Hydroperoxide on Endothelial Dysfunction Through Inhibition of Oxidative Stress and Restoration of eNOS in Rats, Kidney Blood Press Res. 2017; 42: 919-932.
- Sahoo S, Meijles DN, Pagano PJ. NADPH oxidases: key modulators in aging and age-related cardiovascular diseases?, Clin Sci (Lond). 2016; 130: 317-335.
- Cave AC, Brewer AC, Narayanapanicker A, et al. NADPH oxidases in cardiovascular health and disease. Antioxid Redox Signal. 2006; 8: 691-728.
- Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev. 2007; 87: 245-313.
- Martyn KD, Frederick LM, von Loehneysen K, et al. Functional analysis of Nox4 reveals unique characteristics compared to other NADPH oxidases. Cell Signal. 2006; 18: 69-82.
- Lukosz M, Jakob S, Buchner N, et al. Nuclear redox signaling. Antioxid Redox Signal. 2010; 12: 713-742.
- Bhakat KK, Mantha AK, Mitra S. Transcriptional regulatory functions of mammalian AP-endonuclease (APE1/Ref-1), an essential multifunctional protein. Antioxid Redox Signal. 2009; 11: 621-638.
- Ando K, Hirao S, Kabe Y, et al. A new APE1/Ref-1-dependent pathway leading to reduction of NF-kappaB and AP-1, and activation of their DNA-binding activity. Nucleic Acids Res. 2008; 36: 4327-4336.
- Nishi T, Shimizu N, Hiramoto M, et al. Spatial redox regulation of a critical cysteine residue of NF-kappa B in vivo. J Biol Chem. 2002; 277: 44548-44556.
- Seemann S, Hainaut P. Roles of thioredoxin reductase 1 and APE/Ref-1 in the control of basal p53 stability and activity. Oncogene. 2005; 24: 3853-3863.
- Ema M, Hirota K, Mimura J, et al. Molecular mechanisms of transcription activation by HLF and HIF1alpha in response to hypoxia: their stabilization and redox signal-induced interaction with CBP/p300. EMBO J. 1999; 18: 1905-1914.
- Xanthoudakis S, Miao G, Wang F, et al. Redox activation of Fos-Jun DNA binding activity is mediated by a DNA repair enzyme. EMBO J. 1992; 11: 3323-3335.
- Hirota K, Matsui M, Iwata S, et al. AP-1 transcriptional activity is regulated by a direct association between thioredoxin and Ref-1. Proc Natl Acad Sci U S A. 1997; 94: 3633-3638.
- Wei SJ, Botero A, Hirota K, et al. Thioredoxin nuclear translocation and interaction with redox factor-1 activates the activator protein-1 transcription factor in response to ionizing radiation. Cancer Res. 2000; 60: 6688-6695.
- Chen H, Li D, Saldeen T, et al. Transforming growth factor-beta(1) modulates oxidatively modified LDL-induced expression of adhesion molecules: role of LOX-1. Circ Res. 2001; 89: 1155-1160.
- Sakamoto Y, Miyazaki A, Tamagawa H, et al. Specific interaction of oxidized low-density lipoprotein with thrombospondin-1 inhibits transforming growth factor-beta from its activation, Atherosclerosis 183 (2005) 85-93.
- Chen CL, Liu IH, Fliesler SJ, et al. Cholesterol suppresses cellular TGF-beta responsiveness: implications in atherogenesis. J Cell Sci. 2007; 120: 3509-3521.