
Venous Thrombophilia, Platelet von Willebrand Factor Mediated Arteriolar Microvascular Thrombosis in JAK2V617F Mutated Thrombocythemia and Acquired ADAMTS13 Deficiency as Causes of Intrahepatic Obstructive Microvascular Liver Diseases in Budd-Chiari Syndrome and Splanchnic Vein Thrombosis
Jan Jacques Michiels1,3*, Petr Dulicek2, Zwi Berneman1, Alain Gadisseur1, Wilfried Schroyens1
1Departments of Hematology and Coagulation Research, University Hospital Antwerp, Belgium
2Fourth Department of Internal Medicine–Hematology, University Hospital in Hradec Kralove and Charles University Prague, Faculty of Medicine in Hradec Kralove and Prague, Czech Republic on behave of the Central European Vascular Forum: CEVF
3Blood, Coagulation and Cardiovascular Medicine Research Center and International Collaboration and Academic Research on Myeloproliferative Neoplasms: ICAR.MPN, Goodheart Institute & Foundation in Nature Medicine & Health, Rotterdam, The Netherlands
Abstract
Congenital venous thrombophilia is associated with increased risk of venous thrombosis at adolescent and adult age, with recurrent abortions and fetal loss in females, and less frequently with splanchnic vein thrombosis, but not with arteriolar microvascular circulation disturbances. Based on original observations in view of the literature on thrombocythemia in patients with essential thrombocythemia (ET) and polycythemia vera (PV), both ET and PV are associated with increased risk of platelet-von Willebrand factor (VWF) mediated arteriolar microvascular circulation disturbances at adult age, with recurrent abortions and fetal loss in females, and less frequently with splanchnic vein thrombosis, but not with venous thromboembolism.
The high incidences of congenital venous thrombophilic factors including antithrombin III, protein C and S, factor V Leiden and the prothrombin G20210A mutation, acquired lupus anticoagulant as well as the presence of the JAK2V617F mutation indicative for thrombocythemia in trilinear myeloproliferative disease (MPD) are described as the underlying hypercoagulable states in patients with Budd-Chiari syndrome (BCS) and splanchnic vein thrombosis (SVT). In this editorial we propose the novel concept of coagulation and/or platelet mediated microvascular liver pathology is the primary event for the development of BCS, splanchnic vein thrombosis, and portal hypertension with portal and oesophageal varicosal veins as a serious complication in patients with congenital thrombophilia and/or JAK2V617F mutated sticky platelets in clonal ET and PV. Clinical and liver pathology observations are in line with a two hit hypothesis of coagulation- and/or platelet-mediated thrombosis in the liver microcirculation as the underlying etiology of BCS and splanchnic vein thrombosis in patients with congenital venous thrombophilia and/or an acquired JAK2V617F mutated thrombocythemia in ET and PV patients. Severe ADAMTS13 deficiency in advanced liver cirrhosis is related to the severity of liver cell insufficiency due to the combined ADAMTS13 synthesis defect and autoantibodies against ADAMTS13 thereby explaining the more pronounced ADAMTS13 deficiency as compared to the degree of AT III synthesis deficiency in advanced liver cirrhosis. An imbalance between the severely decreased ADAMTS13:AC level and its substrate may indeed reflect the predisposing state for platelet thrombi in the liver microcirculation in patients with advanced liver cirrhosis similar on op of congenital venous thrombophilia and platelet-VWF mediated arteriolar microvascular thrombosis in JAK2V617F mutated thrombocythemia as etiological risk factors of intrahepatic microvascular obstructive diseases in BCS followed by splanchnic vein thrombosis.
Introduction
Hereditary thrombophilia caused by congenital deficiency of an anticoagulant factor or an increased procoagulant factor is associated with an increased risk of coagulation mediated venous thrombosis (figure 1)1. Deficiency of the main physiological anticoagulant factors include deficiency of antithrombin III (AT) Protein C (PC) and Protein S (PS), and the Factor V Leiden (FV Leiden). Increased Factor II levels due to prothrombin G20210A gene mutation and blood group non-O related elevated Factor VIII levels have been identified as risk factors for venous thrombosis. The congenital thrombophilias AT, PC, PS and FV Leiden are associated with increased risk of venous thrombosis at adolescent and adult age, with recurrent abortions and fetal loss in females, and less frequently with splanchnic vein thrombosis, but not with arteriolar microvascular circulation disturbances1. The frequency of hereditary thrombophilia in patients with confirmed idiopathic thrombosis (outside the clinical setting of surgery, trauma, or cancer) is approximately 25%. The most common genetic predisposition to venous thrombosis in Caucasians is activated Protein C (APC) resistance, which is caused by the Factor V Leiden mutation in about 90 %. Combined deficiencies of FV Leiden, PC, PS, AT, the prothrombin mutation G20210A or non-O blood group have been described to lead to a higher risk of venous thrombosis (figure 1)1. The liver produces the pro-coagulant vitamin K dependent procoagulant factors II, VII, IX, X, the vitamin K dependent anticoagulant factors protein C, protein S, and the vitamin K independent to factor FV, FVIII, fibrinogen and antithrombin III2. As AT III is neither vitamin K dependent nor a reactive protein, quantitative measurement of AT III levels using a chromogenic assay best reflect the degree of liver parenchymal function in chronic liver diseases2.
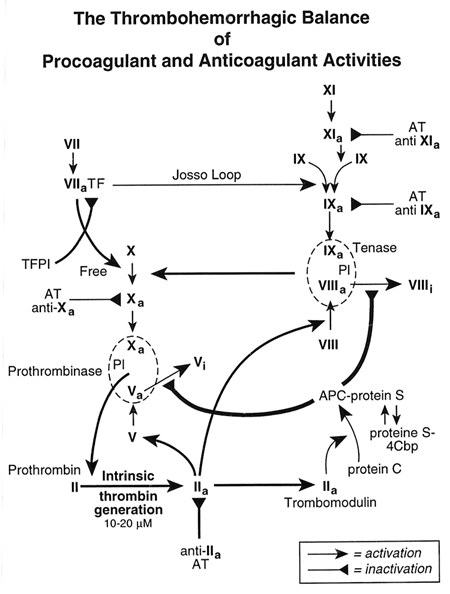
Figure 1: The Thrombohemorrhagic balance of procoagulant and anticoagulant activities in flowing blood designed by Michiels
The PVSG classifications used platelet counts in excess of 600 x109/L as the minimum criterion for the diagnosis of ET3-5, thereby overlooking the early stages of myeloproliferative diseases (MPD). This comprises about 30% of masked MPD, which prompted Michiels & Thiele to define the European Clinical and Pathological (ECP) criteria4. The ECP criteria lowered the platelet count cut-off to 400 x109/L (upper limit of normal) on top of bone marrow histology as a pathognominc clue to the diagnosis of thrombocythemia in various MPDs4. ET according to the ECP criteria comprises three types of WHO defined prefibrotic MPD or myeloproliferative neoplasms (MPNs) including ET, thrombocythemia associated with polycythemia vera (PV), and prefibrotic primary megakaryocytic granulocytic myeloproliferation (PMGM) without features of PV. The ECMP criteria in table 1 include the presence of the JAK2V617F mutation in granulocytes and large clustered megakaryocytes in a normal cellular or hypercellular bone marrow due to increased erythro-megakaryopiesis or erythro-megakaryo-granulopoiesis as pathognomonic clues to the diagnoses of ET and PV (Table 1)3-6. The combined use of JAK2V617F mutation screening and BMB evaluation has the potential to differentiate early stage MPN from reactive thrombocytosis and erythrocytosis with a sensitivity and specificity near to 100% (Table 1)3-6. JAK2V617F mutated thrombocythemia in ET and PV patients is associated with increased risk of platelet-mediated arteriolar microvascular circulation disturbances at adult age, with recurrent abortions and fetal loss in females, and less frequently with splanchnic vein thrombosis, but not with venous thromboembolism7-11. Erythromelalgic microvascular circulation disturbances are caused by aspirin responsive and coumarin resistent platelet-von Willebrand factor (VWF) mediated arteriolar fibromuscular intimal proliferation in JAK2V617F mutated thrombocythemia complicated by VWF-rich arteriolar thrombotic occlusion if left untreated with aspirin7-9. The broad clinical spectrum of microvascular arteriolar circulation disturbances of thrombocythemia in patients with prefibrotic ET and PV (trilinear MPN) include atypical transient ischemic cerebral and ocular manifestations and/or coronary artery disease, and erythromelalgia and its peripheral ischemic complications (Figure 2)7,8. These microvascular disturbances are caused by JAK2V617F mutated hypersensitive platelets, which at high shear stress in arterioles induce von Willebrand factor (VWF) mediated ischemic and thrombotic processes in the arteriolar peripheral, cerebral and coronary circulation and reflect a novel distinct disease entity of aspirin-responsive arterial thrombophilia occuring in about two thirds of ET and PV patients (Figure 2)7-11. Michiels et al discovered in the late 1970s and early 1980s that platelet-VWF mediated areriolar microvascular, ischemic, and thrombotic complications of JAK2V617F mutated thrombocythemia are causally related thrombocythemia in patients with ET and PV associated thrombocythemia. The change of ET into PV during follow-up do aggravate the erythromelalgic and cerebral arteriolar microvascular circulation disturbances into major arterial and venous thrombotic events as the consequence of increased hematocrit, red cell mass (hypervolumemia) and blood hyperviscosity7-11. Correction of the hematocrit to normal (below 0.45) is associated with the relief of major thrombosis but with the persistence of thrombocythemia and its arteriolar microvascular complications, which can best be treated and prevented by low dose aspirin (80 mg once daily) but not by coumadin9-12. The incidence of deep vein thrombosis including splanchnic vein thrombosis in 809 ET patients from 11 retrospective studies reviewed by Griesshammer et al was low, 4% (n=33)11. The European Collaboration on Low-dose Aspirin in PV (ECLAP) study initiated by Landolfi, Michiels and Patrono revealed that in routine daily practice 633 (38.6%) of 1638 PV patients at time of diagnosis already had a history of arterial thrombotic events in 27% and of venous thrombosis in 9%12.
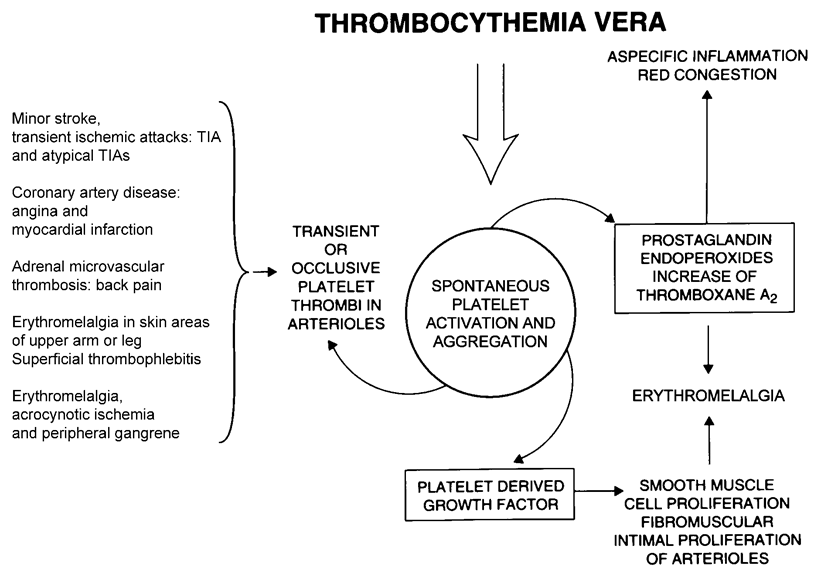
Figure 2: The clinical spectrum of icrovascular circulation disturbances including erythromelalgic, ocular and transient cerebral ischemic manifestations, acute coronary syndrome, microvasculat thrombosis in BCS followedby splanchnic vein thrombosis are causedby platelet mediated thrombotic processes as documented by platelet kinetics and increased thromboxane production during ischemi attacks (Michiels et al PLATELETS 2004;15:67-84, Sem Thromb Hemostas 2006;32:589-604
Congenital Thrombophilia Budd Chiari Syndrome and Splanchnic Vein Thrombosis
Budd-Chiari syndrome (BCS, hepatic vein thrombosis) is a rare disorder, the cause of which remains undetermined in the majority of cases. It is caused by the occlusion of hepatic outflow either at the level of hepatic veins or inferior vena cava (Figure 3 stage 1). BCS has been associated with a variety of conditions like malignancy, polycythemia rubra vera, paroxysmal nocturnal hemoglobinuria, trauma, pregnancy, oral contraceptives, and infection. Deficiencies of congenital anticoagulant factors including antithrombin, protein C, protein S, factor V Leiden, factor II mutation and less frequently acquired lupus anticoagulans or anticardiolipine (ACL) antibodies are well documented underlying causes in about 60% of patients with hepatic vein thrombosis (BCS) and in about 1/3 of patients with portal vein thrombosis (PVT)13-15. Mohanty et al studied the frequency of congenital thrombophilia in 53 BCS and 33 PVT patients in whom MPD was excluded by bone marrow biopsy and in 223 age-matched controls (Figure 4)14. FV Leiden was present in 26% of BCS cases, in 6% of PVT cases and in 2.3% of controls. Antiphospholipid syndrome was present in 21% of BCS and in 18% of PVT cases. The age and gender distribution of BCS and PVT cases show that both BCS and PVT occur between 21 and 50 years with women outnumbering men in that age group 31 to 40 years (Figure 4). Hereditary deficiency of PC, PS or AT was present in 29% of BCS and 13% of PVT patients. Other acquired risk factors like pregnancy, surgery and oral contraceptives were present in 15% of BCS and 9% of PVT patients. In the study of Mohanty 59% of the BCS and 30% of the PVT could be explained by the presence of at least one of the etiologic venous thrombophilic factor. Janssen et al compared 43 BCS and 92 PVT patients with 474 population-based controls in the Leiden Thrombophilia Study15. Among 43 BCS patients MPD was present in 28%, FV Leiden in 26%, hereditary PC deficiency in 9.3%, prothrombin mutation in 4.7% and PS in 0%. Among 92 PVT patients MPD was present in 17%, FV Leiden in 7.6%, hereditary PC deficiency in 6.5%, and prothrombin mutation in 3.2%. The relative risk of BCS was 11.3 for FV Leiden, 6.8 for PC deficiency and 2.1 for prothrombin mutation. The relative risk of PVT was 2.7 for FV Leiden, 4.0 for PC deficiency and 1.4 for prothrombin mutation. Concurrence of either acquired or inherited thrombotic risk factors was observed in 26% of the BCS and in 37% of the PVT patients in the study of Janssen et al15.
A retrospective study of 304 ET/PV patients found Factor V Leiden in 14/304 (4.6%) and MPD was associated with VTE in 5 of 27 (16%) evaluable cases16. The prevalence of Factor V Leiden in these ET/PV patients with and without arterial thrombosis was similar - 5/78 (6%) and 9/211 (4%)16. The prevalence of the allele for Factor V Leiden in another study of 50 MPD patients (17 PV, 15 ET and 18 myelofibrosis (MF) and 30 controls was 9% in the MPD patients and 3.4% in controls17. These two studies suggest that both venous thrombophilia and arterial platelet thrombophilia very likely contribute to increased venous thrombotic risk in MPD (Figures 2 and 3).
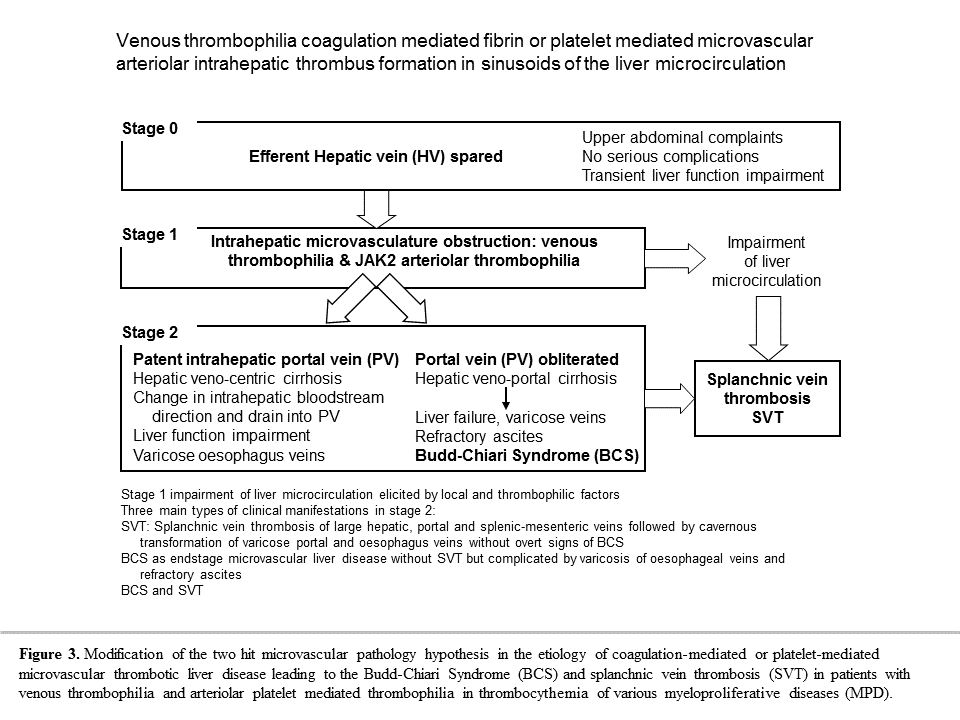
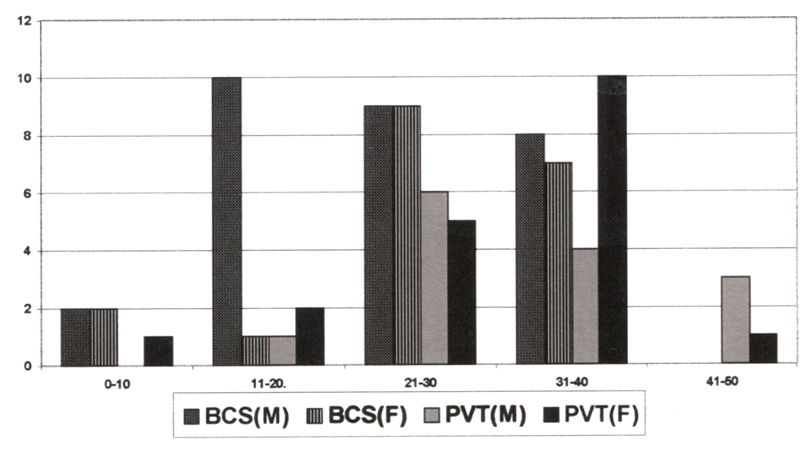
Figure 4:
BCS and Splanchnic Vein Thrombosis in Myeloproliferative Disease (MPD)
Thrombosis in splanchnic veins (hepatic or portal) is rare at time of diagnosis of MPD, but may develop during long-term follow-up18. Thrombosis in splanchnic veins is reported in 19 cases of 460 (4%) consecutive patients with ET19. In one large series of 140 PV patients, thrombosis of major abdominal vessels was observed in 14 (10%)20. A retrospective study of 187 ET patients reported that 60% of all venous thrombosis occurred in either an abdominal vein (n=10) or cerebral sinus (n=2)21. Lengfelder et al reported portal/hepatic/splenic vein thrombosis in 10 of 143 (7%) consecutive ET patients22. In contrast, a chronic MPD of ET, PV or MF can be diagnosed in up to 30% of patients with hepatic vein thrombosis (Budd-Chiari syndrome), and in about 15% to 20% of patients with portal vein thrombosis13,15. Valla et al demonstrated that nearly 40% of patients with splanchnic vein thrombosis (either hepatic or portal) had spontaneous EEC of cultured bone marrow cells as a diagnostic clue for the presence of an underlying overt or latent myeloproliferative disease state23,24. Teofili et al25 assessed spontaneous endogenous erythroid colony (EEC) formation in 43 patients with venous thrombosis before the age of 45 and found that EEC was positive allowing the diagnosis of overt MPD in 4 (2 PV, 1 ET and 1 IMF) and a latent MPD in peripheral blood and bone marrow in 6 patients. All 10 EEC positive suffered from splanchnic vein thrombosis (hepatic 3, portal 2, mesenteric and portal 4, mesenteric 1), but no EEC positivity was found in 25 patients with other sites of deep vein thrombosis (DVT)25. In a review of 120 cases with splanchnic vein thrombosis (Budd-Chiari syndrome 51, and portal/splenic and/or mesenteric vein thrombosis in 69) MPD was diagnosed in 80 by the presence of spontaneous EEC as a clue to MPN in 73 (61%)26. Patients with splanchnic vein thrombosis associated with latent or overt MPD were predominantly females younger than 45 years. The diagnoses according to PVSG criteria in the 80 MPN patients were overt PV in 37 (31%), ET in 2, MF in 2, and latent (masked) MPN in 39 (32.5%)26. From this analysis in 1997 De Stefano, Teofili, Leone & Michiels concluded that both spontaneous EEC and histopathology from bone marrow biopsy provide specific information as sensitive clues for the diagnosis of all variants of latent and overt myeloproliferative disorders26. Using the presence of large clustered megakaryocytes in bone marrow biopsy specimens (table 1), a subsequent French study could diagnose MPN in 46 out of 128 patients with splanchnic thrombosis either hepatic vein or portal vein thrombosis27,28. The sensitivity and specificity of the ECP criteria including bone marrow histopathology to detect masked, early and overt stages of MPN is near to 100% as the underlying etiology in patients with idiopathic splanchnic vein thrombosis (Table 1)4-6. However, the accuracy to diagnose of MPD by the PVSG (1975) and WHO (2001) criteria without the use of bone marrow histology but using clinical criteria of increased red cell mass, low serum EPO levels, EEC, and splenomegaly are insensitive ranging from 52% to 74% to pick up early, masked and latent stages of MPD or MPN4-6,27,28.
Table 1: Diagnosis of the early stage prefibrotic JAK2V617F mutated myeloproliferative disorders essential thrombocythemia (ET), early thrombocythemic prodromal PV and prefibrotic classical PV with trilinear erythrocythemic, megakaryocytic and granulocytic myeloproliferation (EMGM) according to a new set of simplified European Clinical Molecular Pathological (ECMP) criteria4-6
| Clinical and molecular criteria | WHO bone marrow criteria |
|---|---|
| ET | ET |
| 1. Platelet count of >400 x109/l and the presence of large platelets in a blood smear 2. Presence of JAK2V617F mutation 3. Hematocrit: male <0.51, female <0.48, 4. Erythrocytes less than 6.0x1012/L |
Predominant proliferation of enlarged megakaryocytes with hyperlobulated nuclei and mature cytoplasm, lacking conspicuous morphological abnormalities. No increase, proliferation or immaturity of granulopoiesis or erythropoiesis. No or only borderline increase in reticulin. |
| Early prodromal and classical PV | Prodromal and classical PV |
| 1. Platelet count of >400 x109/l 2. Early PV: normal hematocrit: male <0.51, female <0.48, and erythrocytes less than 6.0x1012/L 3. Classical PV: increased hematocrit and erythrocytes above 6.0x1012/L 4. Presence of JAK2V617F mutation 5. Low serum EPO level, increased score for leukocyte alkaline phosphatase (LAP) 6. No or minor splenomegaly |
Increased cellularity due to trilinear erythrocythemic, megakaryocytic, granulocytic myeloproliferation (EMGM = panmyelosis). Proliferation and clustering of small to giant (pleomorphic) megakaryocytes. No pronounced inflammatory reaction (plasmacytosis, cellular debris). Absence bone marrow features consistent with congenital polycythemia and secondary erythrocytosis. No or only borderline increase in reticulin. Spontaneous EEC |
| Prefibrotic EMG trilinear MPN | Prefibrotic EMG (masked PV) MPN |
| 1. Platelet count of >400 x109/l and no signs of leuko-erythroblastosis 2. Slight splenomegaly on ultrasound and no anemia Hb >12g/dl 3. Presence of JAK2V617F mutation 4. No preceding or allied of CML, PV, RARS-T or MDS . |
Erythrocythemic, megakaryocytic and granulocytic trilinear myeloproliferation (EMGM) and relative reduction of erythroid precursors. Dense clustering and increase in atypical giant to medium sized megakaryocytes containing lobulated nuclei and definitive maturation defects. No or only borderline increase in reticulin. |
Patel et al described 41 cases with idiopathic Bud-Chiari syndrome (BCS with absence of congenital thrombophilia in 38), of which 24 carried the JAK2V617F mutation and 17 not, but myeloproliferative neoplasms (MPN) was detected in only 17 cases by clinical criteria without the use of bone marrow histology29. Out of 24 BCS cases positive for the JAK2 mutation, only 10 developed overt PVSG-defined clinical criteria for MPN (7 ET, 3 PV, table 2). Another 4 had evidence of masked MPN (3 ET, 1 PV) at time of presentation. Six of nine presented with splenomegaly and/or a hypercellular bone marrow suspicious for advanced stage of MF. Out of the 17 BCS patients negative for the JAK2V617F mutation, five cases showed evidence for masked MPN on bone marrow histology evaluation (1 developed ET, 1 was polycythemic associated with EEC positivity, 1 showed the combination of EEC positivity, splenomegaly and a hypercellular bone marrow, 2 had splenomegaly and a hypercellular bone marrow29. In four recent studies, the presence of JAK2V617F mutation appeared to be a specific diagnostic clue to myeloproliferative disease in 31% of patients with idiopathic Budd-Chiari syndrome and splanchnic vein thrombosis (table 2)29-32. In two additional reports33,34, patients with splanchnic vein thrombosis, but without signs of overt MPN, the laboratory markers EEC and low serum EPO were less sensitive whereas the combination of JAK2V617F mutation and bone marrow histology assessment was highly sensitive and specific for diagnosis of MPN. Splanchnic vein thrombosis associated with masked or early stage prefibrotic ET was more common in females (table 2)29-34. Kiladjian et al assessed the diagnostic and prognostic value of JAK2 and MPL515 mutations in 241 SVT patients (104 BCS, 137 PVT). JAK2V617F was found in 45% of BCS and 34% of PVT, while JAK2 exon 12 and MPL515 mutations were not detected (figure 5)35. JAK2V617F was found in 96.5% of patients with BM changes specific for MPD and EEC, but also in 58% of those with one feature (BM or EEC), and in 7% of those with neither feature indicating the superiority of JAK2 screening for detection of MPN in SVT patients35. In the meta-analysis of Smalberg et al36, JAK2V617F screening in SVT patients identified underlying MPN disease in 17,1% and 15.4% as could be confirmed by bone marrow histology evaluation36.
Table 2: Detection of JAK2V617F mutation indicative for latent (masked) or overt myeloproliferative disease (MPD) in patients with Budd-Chiari syndrome (BCS, hepatic vein thrombosis) and splanchnic vein thrombosis (SVT) including portal vein thrombosis (PVT) or isolated mesenteric vein thrombosis (MVT)
| Reference Year | Patients N |
JAK2V617F | Overt MPD |
Masked MPD** | |
|---|---|---|---|---|---|
| N | F/M | ||||
| Patel et al29 2006 | 41 BSC | 24 | 16/8 | 0/10* | 13** |
| Smalberg et al30 2006 | 40 BCS | 13 | 9/5 | 13 (WHO) | |
| Collazzo et al31 2007 | 99 PVT | 17 | 11/5 | 7 | 10 |
| De Stefano et al32 2007 | 78 SVT HVT 15- PVT 63 |
32 | 20/12 | 4/15* | 17 |
| Total | 274 | 86 | 56/30 | 37 | 48 |
| BCS/PVT | 100% | 31% | 2 : 1 | 14% | 17% |
* MPD according to WHO criteria became overt during follow-up of SVT
** Masked MPD according to WHO criteria at diagnosis and during follow-up of SVT patients
N =number of patients, F = female, M = male.
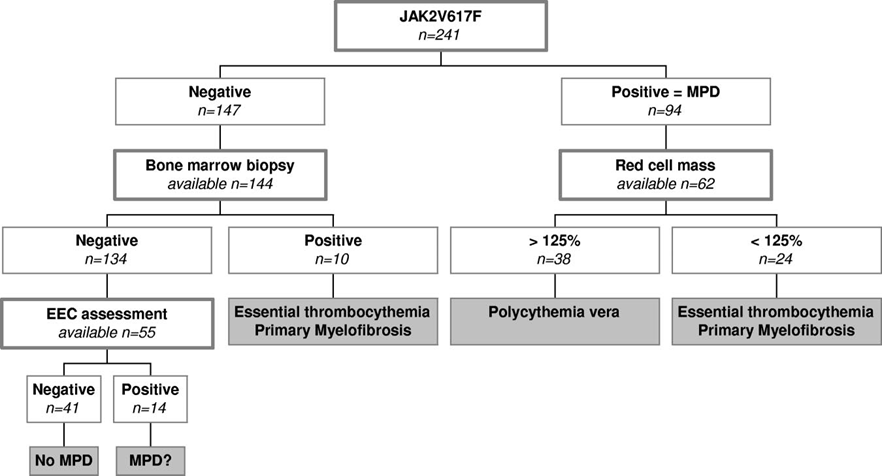
Figure 5:
Non-neoplastic Portal Vein Thrombosis in Liver Cirrhosis
Amitrano et al performed a cross sectional study in a total 701 liver cirrhotic patients diagnosed between 1998 and 2002 with Doppler ultrasonography and found 79 cases (11.2%) with portal vein thrombosis (PTV) usually associated with advanced liver disease37. Of these 79 PVT patients, 34 (43%) were asymptomatic and 45 (57%) were symptomatic and presented with portal hypertensive bleed in 31 (39%), abdominal pain in 14 (18%), and intestinal ischemia or infarction in 10 (12.6%) mainly due to mesenteric vein thrombosis (MVT). Wanless et al analysed 61 cirrhotic livers removed at transplantation to clarify the prevalence, distribution, and pathogenesis of venous lesions, as well as the association of these lesions with other morphological features and clinical morbidity. Wanless et al studied the histopathology of obliterative lesions in intraheptic portal veins (IPV) and intrahepatic hepatic veins (IHV) of all sizes, which are known to occur in cirrhotic livers38. IPV lesions have generally been attributed to thrombosis, but the etiological pathogenesis of the intrahepatic veno?occlusive lesions is unknown. Intimal fibrosis that is highly suggestive of healed IHV or IPV thrombosis was found in at least 70% and 36% of livers, respectively. The distribution of IHV lesions was patchy and largely confined to veins between 0.1 and 3 mm in diameter, suggesting multifocal origin in small veins. IPV lesions were more uniform throughout the liver. IHV lesions were associated with regions of confluent fibrosis (focal parenchymal extinction), and IPV lesions were associated with regional variation in the size of cirrhotic nodules and a history of bleeding varices. These observations suggest that microthrombosis of small to medium IPV and IHV is a frequent occurrence in cirrhosis, and that these events are important in causing progression of cirrhosis complicated by portal hypertension without or with PVT and extended SVT38 (Figure 3 stage 2).
The Concept of BCS Due to Intrahepatic Veno-Portal Obstructive Thrombosis/Cirrhosis
Here we propose the novel concept of intrahepatic veno-portal obliterative microvascular disease related to venous thrombophilia, platelet-VWF arteriolar microvascular thrombosis in JAK2V617F thrombocythemia and ADAMTS13 deficiency as the three main underlying causes of BCS. Budd-Chiari syndrome (BCS: hepatic venous outflow obstruction) and splanchnic vein thrombosis (SVT) has been recognized by many investigators as a microvascular disease caused by a spectrum of underlying disorders including acquired JAK2V617F mutated thrombocythemia in various MPNs, congenital venous thrombophilia including FV Leiden and FII mutations, AT, PC and PS deficiencies, acquired coagulopathies including lupus anticoagulant (LAC), anticardiolopin (ACL) antibodies, paroxysmal nocturnal hemoglobinuria (PNH), and Behcet’s disease, malignancy, surgery and infection13-15,37-39. BCS due to intrahepatic vein thrombosis is an end stage liver disease with severe congestion, fibrosis and cirrhosis. The histological type of fibrosis may range from bridging fibrosis between intrahepatic hepatic veins (IHV) without septal to portal fibrosis to more usual type of cirrhosis with septa bridging of IHV to portal tracts38. Tanaka and Wanless analysed the vascular pathology of 15 explanted whole livers with BCS and correlated histological patterns of cell loss and fibrosis with the distribution of IHV and intrahepatic portal vein (IPV) obstruction39. In the first stage of veno-centric cirrhosis or congestive cirrhosis show early lesions of severe cell loss near hepatic veins (IHV), whereas intrahepatic portal veins (IPV) were usually patent and dilated (figure 3). The peculiar lesion of veno-centric cirrhosis is seldom seen in other diseases, and is seen in pure form in the minority of BCS livers that had not suffered from portal vein thrombosis (figure 3). In the second stage of old lesions, the portal tracts are incorporated into fibrous septa giving veno-portal cirrhosis with invisuable hepatic veins and obliterated portal vein. Some explanted BCS livers had regions of both veno-centric and veno-portal cirrhosis. Areas of veno-centric fibrosis had less hepatic vein (IHV) disease and areas of vena-porta cirrhosis had more severe portal vein (IPV) disease. The large hepatic veins were normal in nearly all BCS patients, whereas bleeding varices before liver transplantation was associated with moderate to severe IPV disease (figure 3). The proposed two hit microvascular pathology hypothesis in BCS illustrates the full range and great heterogeneity of obstruction seen in microvascular obstructive disease and the co-existence of intrahepatic IHV and IPV thrombosis (figure 3)41. In addition, infarcts with contiguous necrosis involving periportal as well as perivenular tissue were noted in livers with combined intrahepatic IHV and IPV obstruction. Acute infarcts were seen in 3 cases, always in association with acute but healed IPV thrombosis, and may be indication for a platelet-mediated obstruction in the end-arterial liver circulation (figures 2 and 3). Based on the concept proposed by Tanaka and Wanless in figure 3, the main clinical manifestations of the intrahepatic venous outlet obstruction as a vascular liver disease in patients with congenital thrombophilia or acquired arteriolar thrombosis in myeloproliferative thrombocythemia are variable and include: 1) SVT the first presenting symptom without overt signs of BCS and complicated by BCS during long-term follow up, 2) various clinical and intrahepatic stages of BCS or intrahepatic venous outlet obstruction recently recognized as a microvascular liver disease complicated by varicosis of oesophageal veins and refractory ascites, but without SVT, and 3) BSC complicated by SVT (figure 3).
The demonstration in 17 BCS patients, with underlying thrombophilia in 5 cases, MPD in 11 cases or combined in 3, that pretransplant vascular imaging showed decreased portal perfusion in 16, and increase arterial perfusion in 9 and post-transplant obstructive intrahepatic portal venopathy and nodular regenerative hyperplasia in all are in line with the concept that intrahepatic BSC is a microvacular veno-portal obstructive liver disease40. In 282 BCS patients only 42 patients (15%) had combined intrahepatic BCS and portal vein thrombosis PVT)41. BCS without PVT or SVT thrombosis is usually complicated by varicosis of oesophageal veins and ascites due to intrahepatic obstruction (figure 3). All patients with BCS-PVT had occlusion of hepatic and portal veins. Eighteen showed additional splenic vein (N=8), superior mesenteric vein (n-2) or both (N=8) thrombosis and 1 exhibited thrombosis of the inferior caval vein. The underlying disorders in 33 evaluable BCS-PVT patients were both myeloproliferative neoplasm (MPN) including PV, ET, MF and unclassifiable MPN in 9, as well as congenital thrombophilia in 15 and paroxysmal nocturnal hemoglobinuria (PNH), Behcet’s disease or oral contraceptive use in 8 and support the concept of coagulation and platelet mediated etiology of BSC and SVT (Figures 1, 2 and 3)42. The number of etiological venous or arteriolar thrombophilic factors and eliciting local or other risk factors increased significantly with the extent of splanchnic vein thrombosis42. All these clinical data and liver pathology observations are in line with the two hit hypothesis of procoagulant-mediated fibrin and/or JAK2V617F mutated platelet-mediated arteriolar thrombosis in the liver microcirculation in patients with congenital and acquired procoagulant thrombophilia and combined procoagulant- and platelet-mediated microvascular in JAK2V617F mutated clonal thrombocythemia in ET and PV patients (Figures 1, 2 and 3). A search for the presence congenital and acquired thrombophilic risk factors for venous thrombosis, and JAK2V617F mutation screening as a specific clue for sticky platelets in clonal thrombocythemia is warranted in patients with a first episode of BCS and splanchnic vein thrombosis. Subsequent bone marrow biopsy evaluation even in the absence of the JAK2V617F mutation is indicated to rule in or out latent, masked and overt stages of MPN into ET, PV and MF (table 1).
Low molecular weight heparin followed by vitamin K antagonist anticoagulation is the treatment of choice of splanchnic vein thrombosis (SVT). Variables of prognostic significance in a large study of 172 patients with portal vein thrombosis (PVT) were hepatic disorders in 30%, abdominal inflammation in 17%, malignancies in 24% abdominal intervention in 23% hypercoagulability in 27% and MNP in 14%43. In the absence of cancer, liver cirrhosis and mesenteric thrombosis, the survival of PVT without BCS was 95%, 89% and 81% after 1, 5 and 10 years follow-up43. The presence of MPN did not influence survival41. In a cohort of 136 non-malignant non-cirrhotic PVT patients, of whom 84 received anticoagulants the underlying causes of extrahepatic PVT were MPN in 31%, antiphospholipid syndrome in 17%, congenital thrombophilia in 165, septic phlebitis in 11%, other causes in 16% and no causes in 28%43. Thirty-eight thrombotic events in 26 patient occurred during 691 patient years follow-up (5.5 per 100 patient years) as DVT in 18, pulmonary embolism in 5, mesenteric thrombosis in 8 and arterial thrombosis in 543. Underlying prothrombotic state including congenital venous thrombophilia and MPN (PV, ET or MF) and absence of anticoagulant treatment were independent predictors for thrombosis. The studies of Van Genderen et al and Landolfi et al produced good evidence that anticoagulation combined with low dose aspirin prevents arteriolar microvascular thrombosis including BSC and PVT in MPN patients with symptomatic essential thrombocythemia (ET) or polycythemia vera (PV)44,45. Hoekstra et al from the Erasmus University Medical Center, Rotterdam studied 44 patients with PVT shown in figures 6 and 7 and searched for an underlying MPN (Chief JJ Michiels & F Leebeek 1985-2018)46. Based on clinical presentation and results of imaging 13 patients (30%) presented with acute PVT and 31 patients had already signs of portal hypertension (gastrointestinal varices, bleeding and splenomegaly) consistent with chronic PTV. In 31 patients (70%) PVT was the initial diagnosis of MPN. In the majority of these cases laboratory values around the time of PVT diagnosis were not suggestive for an underlying MPN according to WHO criteria, but MPN was confirmed by bone marrow histology in all. The JAK2V617F mutation was present in 26 of 29 tested MPN patients. The overall survival of 44 patients is shown in figure 7 of whom 31 were MPN cases, 20 had another venous thrombophilic factor and 5 cases had two risk thrombophilic factors for PVT. As shown in figure 6, twenty-three (52%) received standard Vitamin K antagonist (VKA) anticoagulation, and long term VKA anticoagulation was given in 15 cases after diagnosis of PVT. Anticoagulation was more frequently given in acute PVT as compared to chronic PVT (77% vs 42% respectively). A final retrospective analysis comparing low dose aspirin on top of VKA or no VKA was associated with no thrombotic events in 12 aspirin treated cases (Figure 6). There were 2 arterial and 1 venous thrombotic events in 9 cases not on aspirin on top of VKA and 2 arterial and 7 venous thrombotic events not on aspirin and VKA (Figure 6). A total of five patients died from end-stage myelofibrosis (2 PV and 3 MF). Survival was not significantly different between patients treated without or with VKA and aspirin (Ascal 100 mg = aspirin 80 mg once daily) did not significantly influence survival (P=0.378) in this small cohort study. Treatment with low dose aspirin (80 mg once daily) seems to us of huge importance in the prevention of all variants of microvascular thrombosis in thrombocythemia of various molecular etiology (Figure 6), but this requires further prospective studies47. Life expectancy and mortality are primarily related to the early stage of underlying MPN and not to complications of PVT (Figure 7)46.
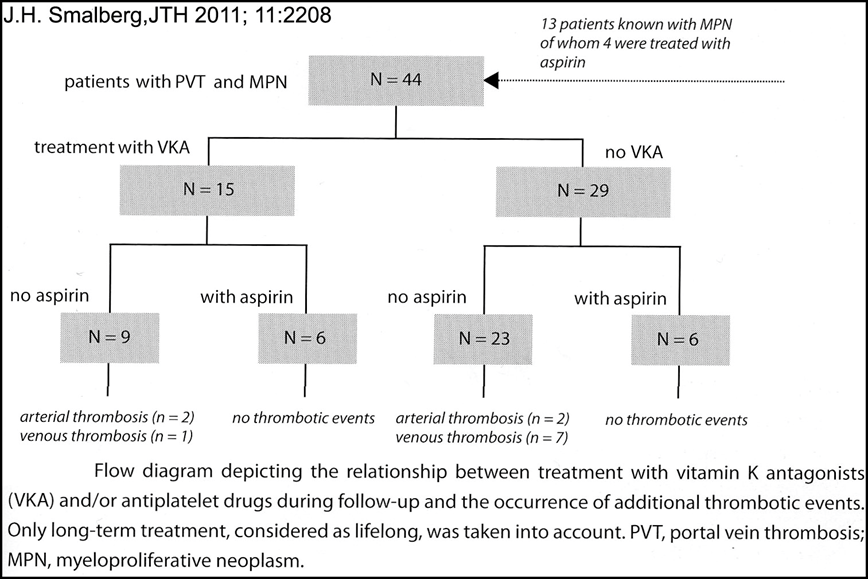
Figure 6:
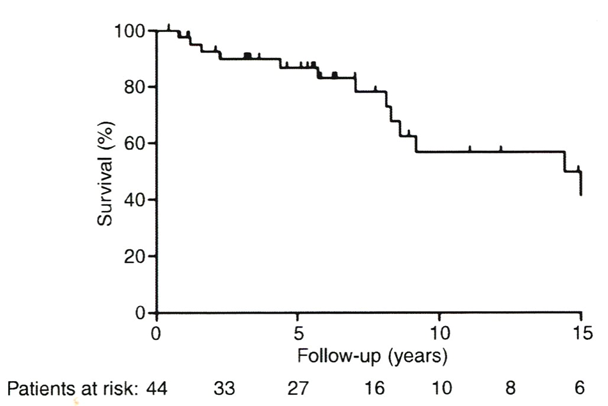
Figure 7:
AT III is a vitamin K independent protein synthesized by liver cells. Quantitative measurement of AT III levels using a chromogenic assay is the best objective marker of the degree of liver paranchymal insufficiency in liver cirrohosis patients2,4. Quantitatively deficient changes in AT III levels are very well known to be associated with the extent of liver cell insufficiency in all variants of liver cirrhosis patients and not influenced by relative or true vitamine K deficency2,4. Bhalli et al studied the role of AT III levels as a nonivasive marker for the degree of histology proven cirrhosis in a well defined group of chronic hepatitis C patiënts48. The mean values of the AT III levels reflecting the liver parenchymal function in stage 0-3 liver fibrosis (N=25) versus stage 4-6 liver fibrosis (N=25) was 96.5 +12% (range 78-117) and 58.9 + 22% (range 23-111) respectively (p-value <0.001, Figure 8)50.
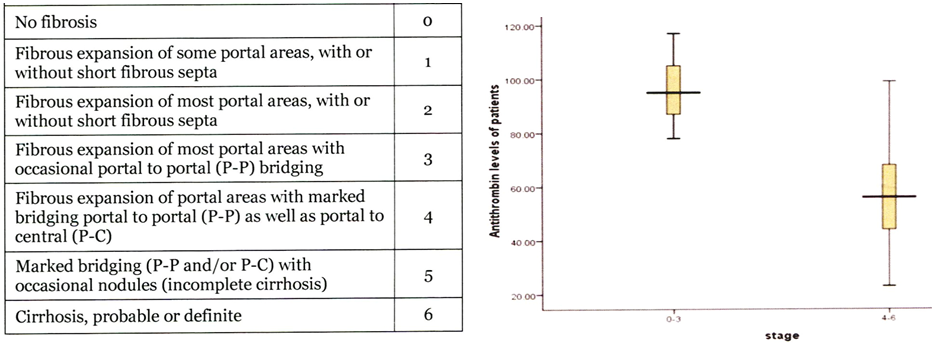
Figure 8:
In the landmark study of Uemara et al the degree of both ADAMTS13 antigen (Ag) and ADAMTS13 Activity (AC depending on the method used) levels are strongly correlated to the degree of liver parenchymal dysfunction as measured by the Child A, B vs C classification (Figure 9)49,50,51. Severe ADAMTS13:AC (<3%) was measured in five liver cirrhosis patients with Child C by the VWFMM assay (Furlan method)52, but the ADAMTS13:AC-ELISA ranged from <5% to 15.9% in the ADAMTS13 AC-ELISA assay49. Interestingly, ADAMTS13 inhibitor was detected in all five liver cirrhosis patients with severe ADAMTS13:AC (Furlan method) (<3%), in 19 of 22 liver cirrhosis patients with moderate (3-25%) ADAMTS13 deficiency and in 4 of 22 liver cirrhosis with mild ADAMTS13 deficiency. In this study, Uemura et al produced good evidence that severe ADAMTS13 deficiency in advanced liver cirrhosis are decreased with increasing severity of cirrhosis due autoimmune antibodies aganinst ADAMTS13 on top of a synthesis defect thereby explaining the more pronounced ADAMTS13 deficiency in advanced liver cirrhosis as compared to AT III synthesis deficiency in liver cirrhosis (Figure 9)49,50. Uemera et al found increased VWF:Ag and VWF:RCo levels, but decreased platelet counts coincidental with severly decreased ADAMTS13:AC, which suggest an additional mechanism for thrombocytopenia related to TTP-like hypercoagulability in patients with advanced liver cirrhosis. Yagita M, Uemura M, Nakamura T et al previously described a case of ADAMTS13 inhibitor development in a case of hepatitis C virus-related liver corrhosis as the cause of thrombotic thrombocytopenic purpura (TTP)51 consistent with the classical diagnosis of TTP (Furlan method)52. ADAMTS13:AC in the case of Yagita et al was extremily low and the inhibitor was positive in both heated plasma and purified IgG consistent with the diagnosis of TTP51. An imbalance between the severely decreased ADAMTS13:AC level and its substrate ADAMTS13:Ag may indeed reflect the predisposing state for platelet-VWF mediated platelet thrombi in the complex liver microcirculation in patients with advanced liver cirrhosis similar as has been observed for anticoagulant responsive congenital and acquired venous thrombophilia and aspirin responsive platelet-VWF mediated peripheral, ocular, cerbral and coronary microvascular thrombosis in JAK2V617F mutated thrombocythemia7-11 very likely also play an important etiological role as causative risk factors of intrahepatic microvascular obstructive diseases in BCS followed by SVT49-51. Targeted well designed prospective clinical and basic research and follow-up studies in newly diagnosed BCS, SVT and PVT patients are warranted within a novel setting of clinical, laboratory, pharmacological and molecular biology in nature medicine.
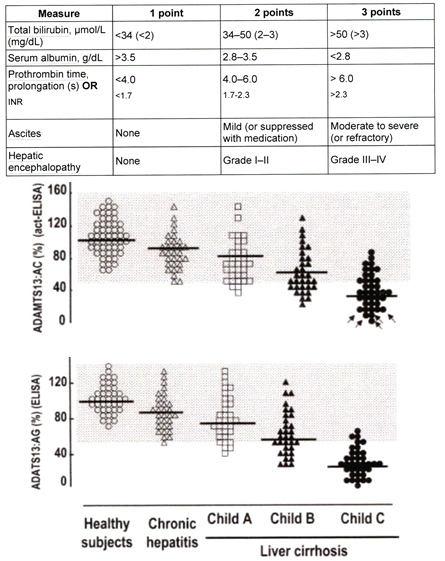
Figure 9:
Declaration of interest
The first author founded in 2000 the Goodheart Institute & Foundation in Nature Medicine & Health, Rotterdam, The Netherlands, Freedom of Science and Education, European Free University Network. JJ Michiels is co-founder of the Central European Vascular Forum (CEVF) and serves as consultant professor in the Bloodcoagulation, Hemostasis Research Laboratory (co-founder VWF-VWD and MPN research programs) at the department of Hematology University Hospital, Antwerp; as consultant to the Dutch Society of Internal Medicine and Ministery of Public Health; consultant of R&D quality driven Industrial and Pharmaceutical Medicine; as an editor of 3 Medical Journals and as a guest editor on request and by self initiation.
Acknowledgement
The authors are grateful to Dr Frank Leebeek Erasmus University Medical Center, Rotterdam for his significant contributions to the literature on Thrombophilia in BCS.
References
- Nicolaides N, Fareed J, Kakkar AK, et al. Prevention and treatment of venous thromboembolism. International Consensus Statement. Int Angiol. 2005; 24(1): 1-26.
- Tripodi A, Mannucci PM. The coagulopathy of chronic liver disease. N Engl J Med. 2011; 365: 147-156.
- WHO classification of the chronic myeloproliferative diseases (CMPD) polycythemia vera, chronic idiopathic myelofibrosis, essential thrombocythemia and CMPD unclassifiable. In: Jaffe S, Harris NL, Stein H et al, editors. WHO Classification of Tumours. Tumours of Haematopoiesis and Lymphoid Tissues. Lyon. IARC. 2001; 31-42.
- Michiels JJ, Thiele J. Clinical and pathological criteria for the diagnosis of essential thrombocythemia, polycythemia vera and idiopathic myelofibrosis (agnogenic myeloid metaplasia). Int J Hematol. 2002; 76: 133-145.
- Michiels JJ, Berneman Z, Schroyens, et al. WHO bone marrow features and European clinical molecular and pathological criteria for the diagnosis of myeloproliferative disorders. Leukemia Research. 2007; 31: 1031-1038.
- Michiels JJ, De Raeve H, Berneman Z, et al. The 2001 World Health Organization and updated 2006 European Clinical and Pathological criteria for the diagnosis, classification and staging of Philadelphia-chromosome negative chronic myeloproliferative disorders. Sem Thromb Hemostas. 2006; 32: 307-340.
- Michiels JJ, Berneman Z, Schroyens W, et al. Platelet-mediated thrombotic complications in patients with ET: reversal by aspirin, platelet reduction, and not by coumadin. Blood Cells Molecules & Diseases. 2006; 36: 199-205.
- Michiels JJ, Berneman Z, Schroyens W, et al. Platelet-mediated erythromelalgic, cerebral, ocular and coronary microvascular ischemic and thrpmbotic manifestations in patients with essential thrombocythemia and polycythemia vera: a distinct aspirin-responsive and coumadin-resistent arterial thrombophilia. PLATELETS. 2006; 17(8): 528-544.
- Michiels JJ, Berneman Z, Schroyens W, et al. Aspirin-responsive painful red, blue, black toe, or fingers syndrome in polycythemia vera associated with thrombocythemia. Ann Hematol. 2003; 82: 153-159.
- Landolfi R, Marchioli R, Kutti J, et al. Efficacy and safety of low-dose aspirin in polycythemia vera: results of the ECLAP trial. N Eng J Med. 2004; 350: 114-124.
- Griesshammer M, Bangerter M, van Vliet HHDM, et al. Aspirin in essntial thrombocythemia: status quo and quo vadis. Sem Thromb Hemostas. 1997; 23: 371-377.
- Marchioli R, Finazzi G, Landolfi R, et al. Vacular and neoplastic risk in a large cohort of patients with polycythemia vera. J Clin Oncol. 2005; 23(10): 2214-2232.
- Denninger MH, Chait Y, Casadval N, et al. Cause of portal or hepatic vein thrombosis in adults: the role of multiple concurrent factors. Hepatology. 2000; 21: 587-591.
- Mohanty D, Shetty S, Ghosh K, et al. Hereditary thrombophilia as a cause of Budd-Chiari syndrome: a study from Western India. Hepatology. 2001; 34: 666-670.
- Janssen HL, Meinardi JR, Vleggaar FP, et al. FactorV Leiden mutation, prothrombin gene mutation, and deficiencies in coagulation inhibitors associated with Budd-Chiari syndrome and portal vein thrombosis: results of a case-control study. Blood. 2000; 96: 2364-2368.
- Ruggeri M, Gisslinger H, Tosetto A, et al. Factor V Leiden mutation carriership and venous thromboembolism in polycythemia vera and essential thrombocythemia. Amer J Hematol. 2002; 71: 1-6.
- Jensen MK, de Nully Brown P, Thorsen S, et al. Frequency occurrence of anticardiolipin antibodies, Factor V Leiden mutation, and perturbed endothelial function in chronic myeloproliferative disorders. Amer J Hematol. 2002; 69: 185-191.
- Elliot MA, Tefferi A. Thrombosis and haemorrhage in polycythemia vera and essential thrombocythemia. Br J Haematol. 2005; 128: 275-290.
- Gangat N, Wolanski AP, Tefferi A. Abdominal vein thrombosis in essential thrombocythemia: prevalence, clinical correlates, and prognostic implications. Eur J Haematol. 2006.
- Anger B, Haug U, Seidler R, et al. Polycythemia vera. A study of 141 patients. Blut. 1989; 59: 493-500.
- Bazzan M, Tamponi G, Schinco P, et al. Thrombosis-free survival and life expectancy in 187 consecutive patients with essential thrombocythemia. Ann Hematol. 1999; 78: 539-543.
- Lengfelder E, Hichhaus A, Kronewitter U, et al. Should a platelet count of 600 x109/L be used as a diagnostic criterion in essential thrombocythemia? An analysis of the natural course including early stages. Br J Haematol. 1998; 100: 15-23.
- Valla D, Casadeval N, Lacombe C, et al. Primary myeloproliferative disorder and hepatic vein thrombosis. A prospective study of erythroid colony formation in vitro in 20 patients with Budd-Chiari syndrome. Ann Intern Med. 1985; 103: 329-334.
- Valla D, Casadevall N, Huisse MG, et al. Etiology of portal vein thrombosis in adults. A prospective evaluation of primary myeloproliferative disorders. Gastroebeterology. 1988; 94: 1063-1069.
- Teofili L, De Stefano V,, Leone G, et al. Hematological causes of venous thrombosis in young people; high incidence of myeloproliferative disorders as underlying disease in patients with splanchnic vein thrombosis. Thromb Haemostas. 1992; 67: 2970301.
- De Stefano V, Teofili L, Leone G, et al. Spontaneous erythroid colony formation as the clue to an underlying myeloproliferative disorder in patients with Budd-Chiari syndrome or portal vein thrombosis. Sem Thromb Hemostas. 1997; 23: 411-418.
- Chait Y, Condat B, Cazals-Hatem D, et al. Relevance of the criteria commonly used to diagnose myeloproliferative disorders in patients with splanchnic vein thrombosis. Br J Haematol. 2005; 129: 553-560.
- Brière J. Budd-Chiari syndrome and portal vein thrombosis associated with myeloproliferative disorders: diagnosis and management. Sem Thromb Hemostas. 2006; 32: 208-218.
- Patel RK, Lea NC, Heneghan A, et al. Prevalence of the activating JAK2 tyrosine kinase mutation V617F in the Budd-Chiari Syndrome. Gastroenterology. 2006; 130: 2031-2038.
- Smalberg JH, Murad SD, Braakman E, et al. Myeloproliferative disease in the pathogenesis and survival of Budd-Chiari syndrome. Haematologica. 2006; 91: 1712—1713.
- Collaizzo D, Amitrano L, Tiscia L, et al. The JAK2 V617F mutation frequently occurs in patients with portal vein and mesenteric vein thrombosis. J Thromb Haemostas. 2007; 5: 55-61.
- De Stefano V, Fiorini A, Rossi E, et al. Incidence of the JAK2V617F mutation among patients with splanchnic or cerebral venous thrombosis without overt chronic myeloproliferative disorders. J Thromb Haemostas. 2007; 5: 708-714.
- Primignani M, Barosi G, Bergamaschi G, et al. Role of the JAK2 mutation in the diagnosis of chronic myeloproliferative disorders in splanchnic vein thrombosis. Hepatology. 2006; 44: 1528-1534.
- Boissinot M, Lippert E, Girodon F, et al. Latent myeloproliferative disorder revealed by the JAK2V617F mutation and endogenous megakaryocytic colonies in patients with splanchnic vein thrombosis. Blood. 2006; 108: 3323-3324.
- Kiladjian JJ, Cervantes F, Leebeek FWG. The impact of JAK2 and MPL mutations on diagnosis and prognosis of splanchnic vein thrombosis: a report on 241 cases. Blood. 2008; 111: 4922-29. doi:10.1182/blood-2007-11-125328 PMid:18250227
- Smalberg JH, Arends L, Valla DC, et al. Myeloproliferative neoplasms in Budd-Chiari syndrome and portal vein thrombosis: a meta-analysis. Blood. 2012; 120: 4921. doi:10.1182/blood-2011-09-376517 PMid:23043069
- Amitrano L. Risk factors and clinical presentation of portal vein thrombosis in patients with liver cirrhosis J Hepatol. 2004; 40: 736-741.
- Wanless IR, Wong F, Blendis LM, et al. Hepatic and portal vein thrombosis in cirrhosis. Hepatology. 1995; 21(5): 1238-1247.
- Tanaka M, Wanless IR. Pathology of the liver in Budd-Chiari syndromr: portal vein thrombosis and the histogenesis of veno-centric cirrhosis, veno-portal cirrhosis, and large regenerative nodules. Hepatology. 1998; 27: 488-496.
- Cazals-Hatem D, Vilgrain V, Genin P, et al. Arterial and portal circulation and parenchymal changes in Budd-Chiari syndrome: a study in 17 explanted livers. Hepatology. 203; 37: 510-519.
- Murad SD, Valla, D, de Groen PC, et al. Pathogenesis and treatment of Budd-Chiari syndrome combined with portal vein thrombosis. Am J Gastroenterology. 2006; 101: 83-90.
- Janssen HLA, Wijnhoud A, Haagsma EB, et al. Extrahepatic portal vein thrombosis in adults: aetiology and determinants of survival. Gut. 2001; 49: 720-724.
- Condat B, Pessione F, hillare S, et al. Current outcome of portal vein thrombosis in adults: risk and benefit of anticoagulant agent. Gastroenterology. 2001; 120: 490-497.
- Van Genderen PJJ, Prins F, Michiels JJ, et al. Thromboxane-dependent platelet activation in vivo precedes arterial thrombosis in thrombocythemia: a rationale for the use of aspirin as an antithrombotic agent in ET and PV. Brit J Haematol. 1999; 104: 438-441.
- Landolfi R, Marchioli R, Kutti J, et al. Efficacy and safety of low-dose aspirin in polycythemia vera: results of the ECLAP trial. New Eng J Med. 2004; 350: 114-124.
- Hoekstra J, Bresser EL, Smalberg JH, et al. Long-term follow-up of patients with portal vein thrombosis and myeloproliferative neoplasms. J Thromb Haemostas. 2011; 9: 2208-2214. DOI: 101111/j.1538-7836.2011.04484.x.
- Michiels JJ, Commandeur S, Hoogenboom GJ, et al. JAK2V617F positive early stage myeloproliferative disease (essential thrombocythemia) as the cause of portal vein thrombosis in two middle-aged women: therapeutic implications in view of the literature. Ann Hematol. 2007; 86: 743-800.
- Bhalli H, Mohsin S, Saeed T, et al. Antithrombin III as marker of fibrosis in chronic hepatitis. Biomedica. 2015; 31(3): 228-231.
- Cholongitas E, Papatheodoracis GV, Vangeli M, et al. Systemic review. The model for end-stage liver disease shoulr it replace child-Plugh’s classification for assessing prognosis in cirrhosis. Alimentary Pharm Ther. 2005; 22(11-12): 1079-1089.
- Uemura M, Fujimura Y, Matsumoto M, et al. Comprehensive analysis of ADAMTS13 in patients with liver cirrhosis. Throm Haemostas. 2008; 99: 1019-1029.
- Yagita M, Uemura M, Nakamura T, et al. Development of ADAMTS13 inhibitor in a patients with hepatitis C virus-related liver cirrhosis causes thrombotic thrombocytopenic puprpura. J Hepatol, 2005; 42: 418-423.
- Furlan M, Laemmle B. Aetiology and pathogenesis of thrombotic thrombocytopenic purpura and haemolytic uremic syndrome: the role of von Willebrands factor-cleaving protease. Best Pract Res Clin Haematol. 2001; 14(2): 437-454.