
Regulation of SCN5A by Non-coding RNAs in the Brugada Syndrome Context
Houria Daimi1, Diego Franco2*
1Biochemistry and Molecular Biology Laboratory, Faculty of Pharmacy, University of Monastir, Monastir, Tunisia
2Department of Experimental Biology University of Jaen, Spain
Abstract
In the human heart, the action potential (AP) is initiated and maintained thanks to a fast-activating fast-inactivating Na+ current carried by Nav1.5 channels. The pivotal physiological role of Nav1.5 in the heart is reflected by the important consequences of its coding SCN5A gene mutations. These mutations may lead to an impaired functional expression (including expression level, subcellular localization, trafficking, and/or current density), and are generally correlated with severe cardiac rhythm disorders such as Long QT (LQT) and Brugada syndrome (BrS). In BrS, loss of function mutations in SCN5A account for 35-40% of clinically affected patients and around 400 mutations in the SCN5A gene were identified in probands with BrS. Emerging electrophysiological techniques such as patch clamp along with transgenic animal technologies improved our understanding of the pathogenic mechanisms underlying BrS due to SCN5A variants. However, despite significant advances in defining the pathophysiology of Nav1.5, the molecular mechanisms underlying its regulation and contribution to the disease are poorly understood. It is well established that functional expression of Nav1.5 may be under modulation by post-transcriptional regulators, defining thus its transcript levels in the cell and also the penetrance on its associated diseases. Recently, non-coding RNA (ncRNAs) molecules have been identified as key transcriptional regulators of SCN5A expression in the heart. The present mini-review provides a summary of the role of ncRNAs, especially microRNAs (miRNAs), in the regulation of SCN5A. It mainly focuses on their role in the BrS context and discusses the recent updates and the major gaps still to be elucidated.
Abbreviations
AP: Action potential; LQT: Long QT; BrS: Brugada syndrome; ncRNAs: non-coding RNA; miRNAs: microRNAs; lncRNAs: long non coding RNAs; 3′UTR: 3′ untranslated region; SNPs: Single-nucleotide polymorphisms.
Introduction
The tetrodotoxin (TTX) resistant isoform Nav1.5, encoded by the SCN5A gene, is the predominant isoform of voltage-gated sodium (Na+) channels in the heart1. This channel plays a key role generating the cardiac action potential and maintaining the rapid conduction of electrical signals through cardiac tissues2. Its involvement in the aetiology of numerous cardiac physiopathologies strongly suggests that proper regulation of cell biology and function of the channel is critical for normal cardiac function3. The physiological importance of Nav1.5 in the heart is further reflected by the important consequences of variants (mutations) in its coding gene SCN5A4. Regardless their position in the gene sequence, these mutations may lead to an impaired functional expression (including expression level, subcellular localization, trafficking, and/or current density), and are generally correlated with severe cardiac rhythm disorders such as Long QT (LQT) and Brugada syndrome (BrS)5,6.
Long QT syndrome is a primary electrical disease characterized by prolongation of the repolarization process reflected on the electrocardiogram (ECG) as a prolongation of the QT interval. The disease is associated with potentially lethal ventricular arrhythmias. Genetically, LQT is classified into 17 different genetic subtypes including the SCN5A related type (LQT3) which represents up to 10% of LQT reported cases. Nav1.5 mutations in LQT3 display a gain-of-function either by a pathological increase in INaL or window current or both7.
Brugada syndrome is an arrhythmic disorder characterized by a coved type ST-elevation and T wave inversion in the right precordial lead V1 and/or V2 of the ECG consistent with the type1 Brugada ECG pattern and is associated with an increased risk for ventricular fibrillation and sudden cardiac death8. BrS is inherited on an autosomal dominant pattern and pathogenic variants in SCN5A are beyond 40% of the pathophysiological substrate of BrS in the affected patients9. In some of the cases who tested negative for SCN5A disease-causing variants using conventional sequencing methods, large-scale genomic imbalances (Copy Number Variants, CNVs) have been identified10. SCN5A mutations in the BrS context are most likely associated with Nav1.5 loss-of-function either by decreased expression of Nav1.5 in the sarcolemma, expression of non-functional channels or altered gating properties leading to a decreased INa (e.g., delayed activation or earlier or faster inactivation)11. With the recent advances in molecular biology, it became well established that functional expression of Nav1.5 is also under modulation of post-transcriptional regulators, defining its cellular transcript levels and the penetrance of its associated diseases, particularly BrS12. However, given the complex nature of this sizable gene (nearly 30 exons spanning > 80 kb), SCN5A transcript regulatory mechanisms are far from being totally deciphered although the relentless efforts in the field. An important mechanism affecting the functional expression of SCN5A could be the alternative splicing of wild type and/or mutant Nav1.5 transcripts. Alternative splicing of Nav1.5 creates a total of nine transcripts (Nav1.5a, Nav1.5b, Nav1.5c, Nav1.5d, Nav1.5e, Nav1.5f, truncated variants E28B-D) differentially expressed in the mammalian myocardium1 and in various other tissues including brain, neuronal cell lines13,14, dorsal root ganglia (DRG)15,16, gastrointestinal tract17 and various tumor tissues and cell lines18,19. In the heart, functional consequences of Nav1.5 splicing are diverse. Spliced channels can show altered kinetics (Nav1.5a, Nav1.5d, Nav1.5e), they can be non-functional (Nav1.5b, Nav1.5f, truncated variants E28B-D), or their electrophysiological properties can be virtually unchanged (Nav1.5a, Nav1.5c), when compared to non-spliced Nav1.51. Although alternative splicing is demonstrated to be a potential mechanism that allows generating a variety of functionally distinct Nav1.5 channels, yet scarce information is known on the regulation of the expression and function of all five functional Nav1.5 variants in different heart regions, at distinct developmental stages, and in the diseased heart. In addition, the regulatory mechanisms of SCN5A/Nav1.5 seem to be even more complex with the discovery of non-coding RNAs (ncRNAs) and their wide implication in multiple disease states, including cardiac arrhythmogenic pathologies. Recently, ncRNAs molecules have been introduced as key regulators of SCN5A expression in the heart20.
Non-coding RNAs are commonly considered as RNA transcripts that, although not having a direct protein-coding potential, are biologically functional RNAs with fine-tuned mechanisms of protein-coding gene regulation21. Considering the complexity of such ncRNAs in structures, genomic orientation, function, cellular localization, or other emerging criteria, a simple and common ncRNAs classification was raised22. The most well-accepted classification model was based on the length of RNA: small non coding RNAs (shorter than 200 nucleotides in length) and long non coding RNAs (lncRNAs) (longer that 200 nucleotides in length). Small non-coding RNAs contain distinct subclasses such as piwi-RNAs, snoRNAs, siRNAs, and the most numerous and well-studied group of microRNAs23,24.
MicroRNAs are small non-coding RNAs of 22–24 nucleotides that are capable of regulating gene expression via translational repression and mRNA degradation predominantly by binding to the 3′ untranslated region (3′UTR) of specific mRNAs25,26. lncRNAs consist of at least 200 nucleotides with limited or no protein-coding capacity27. Due to their ability to interact with DNA, RNA, and proteins, lncRNAs are able to regulate very diverse cellular processes such as chromatin modification, transcription, post-transcriptional modifications, scaffolding, and post-transcriptional mRNA regulation28,29. This way, lncRNAs regulate a wide range of biological processes through their crosstalk with miRNAs that, in turn, regulate mRNAs30. Since these cross talking molecules are so closely related, abnormal expression of one of them interferes with mRNA expression patterns and vice versa, an alteration of mRNA expression, most likely due to genetic variants (mutation), create a dysregulation of the interfering ncRNAs that can culminate in disease development as it is the case of several cardiac disorders31.
In the present mini-review we will explain the role of ncRNAs, especially microRNAs (miRNAs), in the regulation of SCN5A. We will focus on their role in the BrS context and discuss the recent updates and the major gaps still to be elucidated.
Regulation of SCN5A by ncRNAs
In the whole genome network analysis, SCN5A was one of the targets identified to be highly regulated by ncRNAs, however, microRNAs are so far the only ncRNA category that have been described in association with SCN5A32. In this regard, our group has performed a screening of the microRNAs predicted to target SCN5A (Figure 1). Using HL-1 cardiomyocytes and luciferase assays, we demonstrated that miR-98, miR-106, miR-200 decrease, and miR-219 increase SCN5A expression levels through directly targeting its 3’UTR, while miR-125 and miR-153 indirectly modulate SCN5A expression through an unknown mechanism. Out of these miRNAs, we demonstrated that miR-219 exceptionally upregulate SCN5A causing an increase in the sodium current in vitro and in vivo miR-219 administration was capable of correcting the QRS prolongation induced by flecainide intoxication in mice20. Furthermore, we demonstrated that miR-219 and miR-200 display a complementary regulatory pattern on SCN5A, in line with their developmental expression profiles.
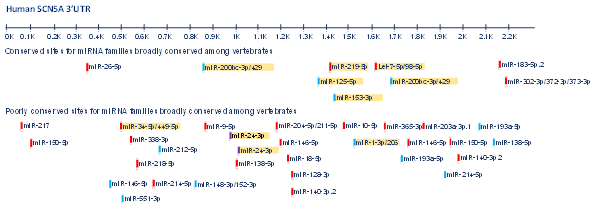
Figure 1: Predicted conserved and poorly conserved microRNA’s binding sites along SCN5A 3’UTR. MicroRNAs highlighted in yellow correspond to those functionally validated as regulators of SCN5A either in vitro or in vivo or both.
A similar study performed by Poon et al. further focused on miR-200c and showed that knockdown or overexpression of this microRNA is associated with changes in SCN5A levels33. Mazzone and co-workers identified four microRNAs with a possible effect on SCN5A. These microRNAs are miR-200a-3p and miR-429 belonging to the miR-200 cluster and Let7-e and Let7-f belonging to the miR-98 cluster. Particularly, let-7f overexpression was shown to decrease SCN5A expression, NaV1.5 current density and electrical excitability in human smooth muscle cells34. Let-7f along with several other microRNAs such as miR-378, miR-125a/b, miR-192-5p, miR-34b/c and miR-1270 are proven to target SCN5A by a direct binding to its 3’UTR and upregulation of these miRNAs is anticipated to cause reduction of INa via downregulating SCN5A/Nav1.5 in the cardiac cells35-38.
Lastly, Yang et al. revealed that miR-1 is able to repress SCN5A, distinctly to our previous data showing that miR-1 over-expression in HL-1 atrial cardiomyocytes resulted in a decrease of GJA1 and KCNJ2 levels but not SCN5A39,40. Thus, additional experiments will be required to solve this controversy.
Role of SCN5A-microRNA crosstalk in Brugada syndrome
Causative variants in SCN5A account for 35–40% of individuals with a clinical diagnosis of BrS41. These variants can be divided into two categories, based on gene location of the variant: SCN5A coding sequence variants, which include rare or common exonic variants and noncoding sequence variants, which include intronic and 3’/5’ UTR noncoding variants42. To date, only rare coding variants in the SCN5A gene have been significantly associated with the syndrome. However, the genotype/phenotype studies conducted in families with SCN5A mutations illustrate the complex mode of inheritance of BrS. This genetic complexity has recently been confirmed by the identification of common polymorphic alleles strongly associated with disease risk43. Even though, noncoding sequence variants remain the major single-nucleotide polymorphisms (SNPs) identified in the disease context, including also BrS44. We previously demonstrated that among the SCN5A variants we have identified in BrS patients, 70% are noncoding genomic variants mainly localized in the 3’UTR45,38.
However, although noncoding sequence variants shape the majority of the BrS genetic background, characterizing their functional effect in the BrS onset remains the major challenge; SCN5A genetic screening and functional characterization either through computational simulation or functional in vitro/in vivo testing generally prioritizes variants in the protein-coding portions and flanking intronic regions of the gene45.
Few emerging studies, including ours, have called the attention to the fact that non-coding sequence variants may modulate the clinical phenotype in BrS by affecting the Nav 1.5 expression level as well as its biophysical properties38,41. Through screening several families affected by BrS, we were able to demonstrate that noncoding SCN5A variants could modulate the expression levels of SCN5A in the affected patients by altering the affinity (decreasing or increasing) of regulatory microRNAs to the 3’UTR of the gene38. In this regard, we have identified through bioinformatics analyses three predicted conserved binding sites for miR-1270 within the SCN5A 3’UTR and demonstrated that the presence of rs4073797 and rs4073796 polymorphisms in the SCN5A 3’UTR may create a fourth binding site for miR-1270 in a genetically conserved region. Our functional assays in HL1 cardiomyocytes demonstrated that miR-1270 overexpression decreases Scn5a expression hypothesizing thus that genetic variants creating a new binding site for miR-1270 in the 3’UTR further decrease SCN5A expression and thus contribute to the genetic bases of BrS.
For long time, gene noncoding sequences were considered as the exclusive binding regions of microRNAs at the target’s transcripts. Coding sequences were often neglected in microRNA-related studies. Recently, Zhang et al. revealed that the terminal coding exon of SCN5A is functionally engaged by miR-24 which exerts a negative control on cardiac SCN5A expression46. Although miR-24 in this study was not investigated in the BrS context, the SCN5A-miR-24 interaction was proved to be altered by the common SCN5A polymorphism D1819D (rs1805126), a polymorphism widely studied in arrhythmic patients particularly those with BrS47,48.
Putative role of lncRNAs in BrS: candidate genes as liaison
As previously said, lncRNAs are able to regulate very diverse cellular processes such as chromatin modification, transcription, post-transcriptional modifications, scaffolding, and post-transcriptional mRNA regulation. Of particular importance is the fact that lncRNAs can modulate the expression and function of microRNAs by acting as endogenous competitive molecules. To date, no lncRNA has been reported to directly or indirectly modulate SCN5A/Nav1.5 expression and/or function. Nonetheless it is important to highlight in this context that other BrS associated genes such as MHY7, IRX5 and CACNA1C have been reported to be regulated by IncRNAs. LncRNA Myheart has been recently reported to be significantly elevated in the blood from AMI patients compared to healthy controls and to regulate MYH6/MYH7 expression by interacting with chromatin remodelling factors in pathological hypertrophy rat models49. CRNDE lncRNA modulates IRX5 expression in distinct human oncogenic processes50-53 by different mechanisms, including regulation of miR-136-5p in hepatocellular carcinoma54. Most importantly KCNQ1OT1 lncRNA modulates miR-384b/CACNA1C expression in an angiotensin II-induced mouse model of atrial fibrillation55 and TCONS_0075467 regulates miR-328/CACNA1C expression in a rabbit atrial fibrillation model56. While no evidence of SCN5A regulation in the BrS context by lncRNAs have yet emerged, these recent reports of the functional role of lncRNAs on BrS associated genes, of particularly importance those of CACNA1C in arrhythmogenic disorders, warrants that their roles in BrS will soon be elucidated.
Conclusions
BrS is characterized by coved-type ST-segment elevation in the right precordial leads (V1-V3)8. Although associated with a typical ECG, the diagnosis of BrS is still challenging. First, due to its complex clinical spectrum specially in presence of asymptomatic patients. At the molecular level, BrS is even more complex. Many candidate genes have been identified so far but still SCN5A is considered the major one (35-40% of the BrS cases)9. Furthermore, the penetrance of BrS is incomplete which make it difficult to establish the genotype phenotype correlation. The incomplete penetrance has been recently attributed to possible transcriptional regulatory mechanisms making a damaging mutation in one BrS patient, tolerated in the others12.
Given the complex nature of SCN5A, it is no surprise to have multilayer transcriptional and post-transcriptional regulatory mechanisms governing its expression. With the discovery of ncRNAs and their wide implication in multiple disease states, including cardiac arrhythmias, more evidences are added to their possible role as key regulators of SCN5A expression in the BrS. Among all ncRNA subclasses, microRNAs are so far the only ones being described as post-transcriptional regulators of SCN5A/BrS either downregulating (eg. miR-200) or upregulating (eg. miR-219) its expression20,32. MicroRNAs target SCN5A by binding to its 3’UTR or even by binding to its coding sequence as in the case of miR-2446. To date, no evidence of SCN5A regulation in the BrS context by lncRNAs have yet emerged however, the reported functional role of lncRNAs on BrS associated genes suggests that the implication of these ncRNAs in BrS is highly possible. In light of these increasingly emerging instances highlighting how ncRNA dysregulation is tightly linked to the pathogenesis of many human cardiac disorders, including BrS, the potential use of ncRNAs as promoting therapeutic targets and diagnostic tools became more and more interesting.
References
- Schroeter A, Walzik S, Blechschmidt S, et al. Structure and function of splice variants of the cardiac voltage-gated sodium channel Na(v)1.5. J Mol Cell Cardiol. 2010; 49(1): 16–24.
- Gavillet, B. Cardiac Sodium Channel Nav1.5 Is Regulated by a Multiprotein Complex Composed of Syntrophins and Dystrophin. Circulation Research. 2006; 99(4): 407–414.
- Abriel H, Kass RS. Regulation of the Voltage-Gated Cardiac Sodium Channel Nav1.5 by Interacting Proteins. Trends in Cardiovascular Medicine. 2005; 15(1): 35–40.
- Kim JB. Channelopathies. Korean J Pediatr. 2014; 57(1): 1–18.
- Tan HL, Bezzina CR, Smits JP, et al. Genetic control of sodium channel function. Cardiovasc Res. 2003; 57(4): 961–973.
- Amin AS, Asghari-Roodsari A, Tan HL. Cardiac sodium channelopathies. Pflugers Arch. 2010; 460(2): 223–237.
- Wilde AAM, Amin AS. Clinical Spectrum of SCN5A Mutations: Long QT Syndrome, Brugada Syndrome, and Cardiomyopathy. JACC Clin Electrophysiol. 2018; 4(5): 569-579.
- Wu CI, Postema PG, Arbelo E, et al. SARS-CoV-2, COVID-19 and inherited arrhythmia syndromes. Heart Rhythm. 2020; S1547-5271(20)30285-X.
- Nielsen MW, Holst AG, Olesen SP, et al. The genetic component of Brugada syndrome. Front Physiol. 2013; 4: 179.
- Mademont-Soler I, Pinsach-Abuin M, Riuro H, et al. Large Genomic Imbalances in Brugada Syndrome. PLoS ONE. 2016; 11(9): e0163514.
- Sieira J, Dendramis G, Brugada P. Pathogenesis and management of Brugada syndrome. Nat Rev Cardiol. 2016; 13(12): 744-756.
- Bezzina CR, Shimizu W, Yang P, et al. Common Sodium Channel Promoter Haplotype in Asian Subjects Underlies Variability in Cardiac Conduction. Circulation. 2006; 113(3): 338–344.
- Yarowsky PJ, Krueger BK, Olson CE, et al. Brain and heart sodium channel subtype mRNA expression in rat cerebral cortex. Proc Natl Acad Sci USA. 1991; 88(21): 9453–9457.
- Frenz CT, Hansen A, Dupuis ND, et al. NaV1.5 sodium channel window currents contribute to spontaneous firing in olfactory sensory neurons. J Neurophysiol. 2014; 112(5): 1091–1104.
- Renganathan M, Dib-Hajj S, Waxman SG. Na(v)1.5 underlies the ‘third TTX-R sodium current’ in rat small DRG neurons. Brain Res Mol Brain Res. 2002; 106(1-2): 70–82.
- Kerr NC, Holmes FE, Wynick D. Novel isoforms of the sodium channels Nav1.8 and Nav1.5 are produced by a conserved mechanism in mouse and rat. J Biol Chem. 2004; 279(23): 24826–24833.
- Osorio N, Korogod S, Delmas P. Specialized functions of Nav1.5 and Nav1.9 channels in electrogenesis of myenteric neurons in intact mouse ganglia. J Neurosci. 2014; 34(23): 5233–5244.
- Zeng D, Kyle JW, Martin RL, et al. Cardiac sodium channels expressed in a peripheral neurotumor-derived cell line, RT4-B8. Am J Physiol. 1996; 270 (5 Pt 1): C1522–C1531.
- Xing D, Wang J, Ou S, et al. Expression of neonatal Nav1.5 in human brain astrocytoma and its effect on proliferation, invasion and apoptosis of astrocytoma cells. Oncol Rep. 2014; 31(6): 2692–2700.
- Daimi H, Estefanía Lozano-Velasco E, Hernández-Torres F, et al. Regulation of Scn5a by microRNAs: miR-219 modulates Scn5a transcript expression and the effects of flecainide intoxication in mice. Heart Rhythm Journal. 2015; 12(6): 1333-42.
- Moazed D. Small RNAs in transcriptional gene silencing and genome defense. Nature. 2009; 457(7228): 413–420.
- Fu XD. Non-coding RNA: a new frontier in regulatory biology Natl Sci Rev. 2014; 1(2): 190–204.
- Lozano-Velasco E, Garcia-Padilla C, Aránega AE, et al. Genetics Of Atrial Fibrilation: In Search Of Novel Therapeutic Targets. Cardiovasc Hematol Disord Drug Targets. 2019; 19(3): 183-194.
- Rynkeviciene R, Simiene J, Strainiene E, et al. Noncoding RNAs in glioma. Cancers Basel. 2018; 11(1).
- Filipowicz W, Bhattacharyya SN, Sonenberg N. Mechanisms of post-transcriptional regulation by microRNAs: are the answers in sight? Nat Rev Genet. 2008; 9(2): 102–114.
- Yamamoto T, Saitoh N. Non-coding RNAs and chromatin domains. Curr Opin Cell Biol. 2019; 58:26–33.
- Batista PJ, Chang HY. Long noncoding RNAs: Cellular address codes in development and disease. Cell. 2013; 152(6): 1298–1307.
- Wilusz JE, Sunwoo H, Spector DL. Long noncoding RNAs: functional surprises from the RNA world. Genes Dev. 2009; 23(13): 1494–504.
- Mercer TR, Dinger ME, Mattick JS. Long non-coding RNAs: insights into functions. Nat Rev Genet. 2009; 10(3): 155–9.
- Dhanoa JK, Sethi RS, Verma R, et al. Long non-coding RNA: its evolutionary relics and biological implications in mammals: a review. J Anim Sci Technol. 2018; 60: 25.
- Chan JJ, Tay Y. Noncoding RNA: RNA regulatory networks in Cancer. Int J Mol Sci. 2018; 19(5): 1310.
- Zhou R, Hang P, Zhu W, et al. Whole genome network analysis of ion channels and connexins in myocardial infarction. Cell Physiol Biochem. 2011; 27(3–4): 299–304.
- Poon ENY, Hao B, Guan D, et al. Integrated transcriptomic and regulatory network analyses identify microRNA-200c as a novel repressor of human pluripotent stem cell-derived cardiomyocyte differentiation and maturation. Cardiovasc Res. 2018; 114(6): 894–906.
- Mazzone A, Strege PR, Gibbons SJ, et al. microRNA overexpression in slow transit constipation leads to reduced NaV 1.5 current and altered smooth muscle contractility. Gut. 2020; 69(5): 868–876.
- Luo X, Zhang H, Xiao J, et al. Regulation of Human Cardiac Ion Channel Genes by MicroRNAs: Theoretical Perspective and Pathophysiological Implications. Cell Physiol Biochem. 2010; 25(6): 571-586.
- Zhao Y, Huang Y, Li W, et al. Post-transcriptional regulation of cardiac sodium channel gene SCN5A expression and function by miR-192-5p. Biochim Biophys Acta. 2015; 1852(10): 2024–34.
- Nassal DM, Wan X, Liu H, et al. KChIP2 is a core transcriptional regulator of cardiac excitability. eLife. 2017; 6: e17304.
- Daimi H, Haj Khelil A, Neji A, et al. Role of SCN5A coding and non-coding sequences in Brugada syndrome onset: What's behind the scenes? biomedical journal. 2019; 42(4): 252 e260.
- Yang, Lu Y, Wang Z. Control of cardiac excitability by microRNAs. Cardiovasc Res. 2008; 79(4): 571-580.
- Chinchilla A, Daimi H, Lozano-Velascoe N, et al. PITX2 Insufficiency Leads to Atrial Electrical and Structural Remodeling Linked to Arrhythmogenesis. Circulation. 2011; 4(3): 269-79.
- Kapplinger JD, Tester DJ, Alders M, et al. An international compendium of mutations in the SCN5A-encoded cardiac sodium channel in patients referred for Brugada syndrome genetic testing. Heart Rhythm. 2010; 7(1): 33–46.
- Savio-Galimberti E, Argenziano M, Antzelevitch C. Cardiac Arrhythmias Related to Sodium Channel Dysfunction. Handb Exp Pharmacol. 2018; 246: 331-354.
- Gourraud JB, Barc J, Thollet A, et al. The Brugada Syndrome: A Rare Arrhythmia Disorder with Complex Inheritance. Front Cardiovasc Med. 2016; 3: 9.
- Zhou J, Troyanskaya OG. Predicting effects of noncoding variants with deep learning–based sequence model. Nat Methods. 2015; 12(10): 931–934.
- Daimi H, Haj Khelil A, Ben Hamda K, et al. Absence of family history and phenotype-genotype correlation in pediatric Brugada syndrome: more burden to bear in clinical and genetic diagnosis. Pediatr Cardiol. 2015; 36(5): 1090e6.
- Zhang X, Yoon JY, Morley M, et al. A common variant alters SCN5A-miR-24 interaction and associates with heart failure mortality. J Clin Investig. 2018; 128(3): 1154.
- Wilde AA, Brugada R. Phenotypical manifestations of mutations in the genes encoding subunits of the cardiac sodium channel. Circ Res. 2011; 108(7): 884e97.
- Ritchie MD, Denny JC, Zuvich RL, et al. Genome- and phenomewide analysis of cardiac conduction identifies markers of arrhythmia risk. Circulation. 2013; 127(13): 1377e85.
- Han P, Li W, Lin CH, et al. A long noncoding RNA protects the heart from pathological hypertrophy. Nature. 2014; 2514(7520): 102-106.
- Ellis BC, Molloy PL, Graham LD. CRNDE: A Long Non-Coding RNA Involved in Cancer, Neurobiology, and Development. Front Genet. 2012; 3: 270.
- Zhang M, Gao C, Yang Y, et al. Long Noncoding RNA CRNDE/PRC2 Participated in the Radiotherapy Resistance of Human Lung Adenocarcinoma Through Targeting p21 Expression. Oncol Res. 2018; 26(8): 1245-1255.
- Yang HY, Huang CP, Cao MM, et al. Long non-coding RNA CRNDE may be associated with poor prognosis by promoting proliferation and inhibiting apoptosis of cervical cancer cells through targeting PI3K/AKT. Neoplasma. 2018; 65(6): 872-880.
- Liu T, Zhang X, Yang YM, et al. Increased expression of the long noncoding RNA CRNDE-h indicates a poor prognosis in colorectal cancer, and is positively correlated with IRX5 mRNA expression Onco Targets Ther. 2016; 9: 1437-48.
- Zhu L, Liu Y, Chen Q, et al. Long-Noncoding RNA Colorectal Neoplasia Differentially Expressed Gene as a Potential Target to Upregulate the Expression of IRX5 by miR-136-5P to Promote Oncogenic Properties in Hepatocellular Carcinoma. Cell Physiol Biochem. 2018; 50(6): 2229-2248.
- Shen C, Kong B, Liu Y, et al. YY1-induced upregulation of lncRNA KCNQ1OT1 regulates angiotensin II-induced atrial fibrillation by modulating miR-384b/CACNA1C axis. Biochem Biophys Res Commun. 2018; 505(1): 134-140.
- Li Z, Wang X, Wang W, et al. Altered long non-coding RNA expression profile in rabbit atria with atrial fibrillation: TCONS_00075467 modulates atrial electrical remodeling by sponging miR-328 to regulate CACNA1C. J Mol Cell Cardiol. 2017; 108: 73-85.