
Commentary: Arrhythmogenic Cardiomyopathy Related DSG2 Mutations Affect Cadherin Binding Kinetics
Volker Walhorn1, Mareike Dieding1, Jana Davina Debus2, Raimund Kerkhoff1, Anna Gärtner-Rommel2, Hendrik Milting2, and Dario Anselmetti1
1Experimental Biophysics, Physics Faculty, Bielefeld University, D-33615 Bielefeld, Germany
2Erich & Hanna Klessmann Institute for Cardiovascular Research and Development, Heart and Diabetes Center NRW,University Hospital of the Ruhr-University Bochum, D- 32545 Bad Oeynhausen, Germany
Abstract
Cadherins are calcium dependent adhesion proteins that bridge the intercellular gap and establish a tight connection to adjacent cells. Desmoglein-2 (DSG2) is a specific cadherin of the cell-cell contact in cardiac desmosomes. Mutations in the DSG2 gene are associated with rare but severe heart muscle diseases such as arrhythmogenic right ventricular cardiomyopathy (ARVC). However, the molecular pathomechanism of many DSG2 mutations is unknown. In our recently published research, we report on the homophilic binding properties of wildtype DSG2 and two mutations thereof. We found that wildtype DSG2 binding kinetics differs significantly from the two analyzed mutations whereas thermodynamic properties such as the difference in Gibbs free energy are virtually unaffected. Here, we comment on (bio-) physical concepts how non-covalent bonds are characterized and we highlight the limitations of these concepts. Furthermore, we consistently link molecular binding properties of DSG2 with the macroscopic stability of cellular networks overexpressing a distinct DSG2 species. Finally, on the basis of DSG2 binding kinetics, we draft a biophysically inspired hypothesis of a molecular pathomechanism leading to ARVC.
Commentary
How do (bio-) molecules interact with each other and how does this interplay steer higher-level systems? These are the driving questions of single molecule biophysics. In every cell virtually countless molecules interact with specific partners controlling and regulating numerous processes such as the immune response, allosteric regulation, protein expression, (cellular) adhesion or the structural integrity of tissue. The underlying concept of host guest interaction is well known since Emil Fischer introduced his key and lock model in 18941. With his seminal work he introduced a completely new perspective on the molecular interplay which drives and controls the machinery of life. Today, we know that this molecular communication is mediated by recognition forces that establish highly specific but short-lived bonds between individual molecules. Generally, these bonds are of non-covalent nature. They are constituted by (multiple) comparable weak (and unspecific) interactions like hydrogen bonds, van-der-Waals and hydrophobic interaction or electrostatic forces between complementary shaped surfaces. The understanding how (weak) molecular forces act on and between (single) molecules is highly relevant for understanding and controlling processes on the molecular scale as well as macroscopic properties. In order to quantify these binding reactions, physics offers the theoretical framework of thermodynamics and statistical mechanics, respectively. In the first, the binding reaction of an ensemble of molecules is described by means of temperature or entropy change and energy flux. Here, the measured observables are averaged over the whole system lacking any information of an individual reaction or specific molecules. In contrast, statistical mechanics follows a bottom-up approach. Numerous individual reactions, which are subjected to statistical fluctuations are measured in order to yield quantities such as the bond kinetics (e.g. bond lifetime) characterizing a binding reaction. Notably, both thermodynamics and statistical dynamics are equivalent as they are inherently connected by the ergodic hypothesis2. Each concept merely offers its own yet limited perspective of the very same system.
In our recent work, we characterized the homophilic binding properties of the cardiac cadherin desmoglein-2 (DSG2) and mutations thereof (p.D154E and p.V392I)3. Mutations in the DSG2 gene are known causes of rare but severe heart muscle diseases such as arrhythmogenic right ventricular cardiomyopathy (ARVC), which is associated with a progressive loss of cardiomyocytes and a fibrofatty tissue replacement, predominantly in the right ventricle. In order to link a potential pathomechanism on the molecular scale to a clinical phenotype we undertook a joint approach on the single molecule and cellular scale, respectively. For the nano-mechanical analysis we expressed recombinant DSG2 in eukaryotic cells and used the purified protein for atomic force microscopy (AFM) based single molecule force spectroscopy (SMFS). We applied forces to individual DSG2 homo complexes to analyse their binding properties at the molecular level (Figure 1). SMFS in complement to assays on the cellular scale appears to be particularly suited to probe binding properties of adhesion complexes, as it mimics a physiological force load while probing the bond’s strength. By this means we could provide a comprehensive thermodynamic and kinetic description of DSG2 dimerization. At the cellular scale, we analysed the structural integrity of cellular networks overexpressing a distinct DSG2 species (wildtype or mutation) in a dispase based cell dissociation assay to explore the functional characteristics of the macroscopic phenotype. In brief, we found that DSG2 homodimerization is highly specific but exhibits only a small difference in Gibbs free energy for all tested species (∇G0≈18 ± 1 KJ mol-1). This finding is consistent with the fact that the strand exchange binding motif is unaffected by the mutations. Briefly, regardless if DSG2 bears the tested mutations or not, the homophilic binding reaction of each analysed species releases the same amount of energy. Still, this finding does not contain any information about the reaction speed. Estimating the rate constants of the association and dissociation reaction, we found that DSG2 wildtype binds and unbinds substantially faster than DSG2 variants p.D154E and p.V392I. Interestingly, these mutations heavily affect the kinetics of DSG2 dimerization in such a way that the bond lifetime is prolonged whereas the binding reaction is retarded.
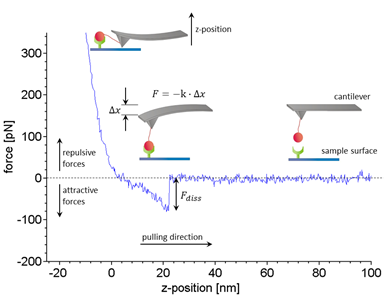
Figure 1:Working principle and typical data of SMFS experiments. Receptors (green) and ligands (red) are covalently bound to a sample surface and a flexible silicon nitride cantilever, respectively (inset). Within a force distance cycle the cantilever is approached towards the sample surface until a certain trigger force is reached and then retracted at a constant velocity. Here, only the retract curve (blue) is shown. In case a ligand binds to a receptor the bond is loaded by an increasing force while the cantilever is deflected successively. The force F acting on the molecular complex is calculated by Hooke’s law as the negative product of cantilever deflection ∇x and spring constant K. The dissociation force Fdiss that was needed to break an individual bond is estimated from the force step that terminates the typical non-linear extension curve. Typically several hundred to thousand dissociation events are needed to characterize a bond. For details of the analysis please refer to our publication3.
In order to quantify the binding characteristics of DSG2 at the cellular scale we tested the resistance of cell networks against external forces in a dispase based cell dissociation assay. Here, cell networks are disrupted by external forces. The structural integrity is estimated by the amount of cell cluster fragments resulting from external mechanic stress. Interestingly, cellular networks overexpressing DSG2 p.D154E exhibit a significantly increased stability. Linking our single molecule and cellular results, we have to consider that quantitative data obtained in affinity assays apply to binding reactions in free solution where unbound molecules can diffuse freely. Within the desmosome however, cadherins are tightly incorporated within the cell membrane through their transmembrane domain whereas diffusion of the free N-terminal end is restricted due to their high density within the desmosome. The bond lifetime of an individual molecular complex is unaffected by this dense configuration. Yet, fast (re-) association is eased as opposing unbound cadherins stay in close proximity after dissociation. Therefore, (free-solution) association rate constants can be estimated as far too small in a spatially confined environment. Keeping this in mind the discrepancy between SMFS and cell assay results solves easily and we can draw a consistent picture: Due to forced re-association, the prolonged bond life time of DSG2 p.D154E results in an increased stability of cellular networks. For DSG2 p.V392I the picture is not that clear. Despite of an impaired binding kinetics the average cell cluster stability is comparable to the DSG2 wildtype. Nevertheless, this finding appears to be in line with the ARVC database classification estimating DSG2 p.V392I as a variant of unknown significance.
These results underline the immense significance of binding kinetics at the single molecule level. In fact, the cardiac desmosome which in the macroscopic picture appears rather static turns out to be a composition of cooperative, highly dynamic constituents whose finely tuned (un-) binding kinetics steer the stability of the whole cellular network. This insight raises the query why nature relies on a configuration of many loosely bound cell-cell contacts in a system that is continuously subjected to alternating forces. Furthermore, it is a matter to explore how an impaired cadherin binding kinetics affects the physiological functionality of cardiomyocytes.
Here, we offer two biophysically inspired suggestions for the pathogenicity of DSG2 p.D154E that are based on DSG2 dimerization kinetics. Firstly, the prolonged bond life time of DSG2 p.D154E homocomplexes might affect the continuous integration and degradation of cadherins within the desmosome. The degradation process might be impaired due to the increased bond lifetime (τ = 3.4 s for p.D154E and τ = 1.7 s for p.V392I), which finally lead to a loss of cardiomyocytes. Consequently, the protein turnover of desmosomal cadherins appears to be of critical relevance in DSG2 related cardiomyopathies. And secondly, the short-living bonds of DSG2 wildtype dimers (average bond lifetime, τ = 0.3 s ) allow a fast realignment DSG2 bonds. Dependent of the deformation of the cell membrane they can rebind to those counterparts that can be reached best. Consequently, this suggests that the cardiac desmosome can adapt its configuration and adhesive properties to an external load. Interestingly, the DSG2 dimer bond lifetime is in the same range as the highest physiological heart beat rate (≈ 180 bpm) allowing the hypothesis that the assumed process of force mediated desmosomal adaption can be accomplished within a heartbeat.
Finally, our results allow a first glimpse into the dynamic interplay of DSG2 within the desmosome and the impact of certain mutations on binding kinetics and cell network stability. Yet, further effort is necessary to gain a consistent picture of desmosomal adhesion. Particularly, the homo- and heterophilic binding of the cardiac cadherin desmocollin 2 and the stability of adhesion clusters that are formed by many parallel bonds have to be in focus.
Financial Support
Financial support by the German Research Foundation (DFG) within the grants DA 370/6-1 to D.A. and HM 1146/2-1 to H.M. is gratefully acknowledged.
The authors declare that they have no competing interests.
References
- Fischer HE, Pinner A. Berichte der deutschen chemischen Gesellschaft. 1894; 27(3): 2643–2658.
- Boltzmann L. Vorlesungen über Gastheorie Band 2. 1898; volume 2.
- Dieding M, Debus JD, Kerkhoff R, et al. Scientific reports. October 2017; 7: 13791.