
Understanding Noncompaction Cardiomyopathy: A Brief Comprehensive Review of A Controversial Entity
Luis Graca Santos*, Rita Ribeiro Carvalho, Sara Fernandes, Joao Morais
Department of Cardiology, Leiria Hospital Centre, Leiria, Portugal
Abstract
Noncompaction cardiomyopathy is a heterogeneous and complex entity characterized by hypertrabeculation, typically of the left ventricle. Uncertainties regarding pathogenesis, classification as primary genetic or unclassified cardiomyopathy, diagnostic criteria, and risk stratification have contributed to fuel the discussion surrounding this disorder. Meanwhile, noncompaction phenotype is thought to be the morphological expression of different underlying pathophysiological mechanisms, genetics, and pathologies. Recent studies suggest that distinguishing genetic from nongenetic causes allows risk stratification and may support clinical management and counselling of patients and their relatives. Additionally, advanced cardiac imaging techniques have demonstrated a complementary role in outcome prediction. The purpose of this review is to provide a brief comprehensive review of this controversial entity.
Introduction
Noncompaction cardiomyopathy (NCCM), first described by Grant in 1926 as “left ventricular noncompaction”1, is a heterogeneous myocardial disorder characterized by prominent trabeculae, intratrabecular recesses and a bilayered myocardium composed of a compacted and noncompacted layer2,3. It has been the subject of several studies and publications, but numerous controversies remain3,4. While NCCM is classified as a distinct primary genetic cardiomyopathy by the American Heart Association5, it is regarded as unclassified familial cardiomyopathy by the European Society of Cardiology6. Indeed, despite being traditionally a familial presentation entity it may occur sporadically, isolated or associated with other congenital defects, affecting only the left ventricle (LV) or both4,7,8. Moreover, noncompaction may also present as a congenital or acquired morphological trait shared by many distinct cardiomyopathies (e.g. hypertrophic (HCM), dilated (DCM))6,9 and may become evident in some pressure/volume overload conditions10,11. In the current paper, we aim to provide a brief and comprehensive review of this controversial disorder.
Pathophysiology
It is postulated that NCCM is the result of abnormal intrauterine heart compaction3,4,13. Apical trabeculations appear around day 26 of gestation and, by the seventh week, both ventricles are similarly and primarily filled with a trabeculation network, with at least twice the thickness of the compact ventricular layer13. This is believed to enhance oxygen and nutrients exchange, before coronary artery development, and to compartmentalize the blood before heart septation. Further on, ventricular thickening and remodelling occurs as the compact layer develops through different processes of proliferation, hypertrophy, and layering, to form the specialized contractile structure14. The molecular and cellular mechanisms involved are not completely understood and it is not clear how the mesh of ventricular trabeculations contributes to the compact layer development13,14. However, several signalling pathways (e.g. NOTCH) involving many genes suspected to contribute to this process have been described, specifically BMP10 (bone morphogenic protein-10) and Nkx2.5 that, when knocked out in mice, are associated with hypertrabeculation15-17. Moreover, it was speculated that hypertrabeculation may result from a compensation mechanism of impaired attempt of myocardial growth and hypertrophy: abnormal cardiomyocyte adhesion due to gap junction malfunction or altered endocardium signalling leading to persisting sinusoids penetrating the ventricular wall18.
Nonetheless, some findings oppose this “embryogenic hypothesis”15,18. In fact, hypertrabeculation may also develop during adulthood in a normally compacted LV as an adaptive response to abnormal loading conditions (e.g. athletes, pregnancy, anaemia): a more trabeculated ventricle allows higher stroke volumes with lower wall stress7,19,20. Gap junction malfunction due to intraventricular pressure and microinfarcts secondary to vascularization mismatch in a compensating hypertrophic ventricle are mechanisms that may contribute to acquired hypertrabeculation19,21.
Genetics
Genetics plays an important role in NCCM since in up to 50% of patients there is evidence of a genetic cause, whether because a cardiomyopathy mutation is found and/or because at least one family member presents non-ischemic cardiomyopathy9,22,23.
The genetic causes are heterogeneous but share a final common pathway, similar to other types of cardiomyopathy with multiple causes and several mechanisms at molecular and cellular levels3,8,16. In the familial form, autosomal dominant transmission (with reduced penetrance) is the most common mode of transmission7,22, followed by X-linked, mitochondrial inheritance and chromosomal abnormalities8,24,25. In a systematic review of 541 patients, van Waning et al.25 showed that autosomal dominant inheritance (mostly missense mutations) was the most frequent pattern of transmission (83%), with more than half of the genetic defects being reported in sarcomere genes (MYH7, MYBPC3, ACTC1, and TTN). These mutations were usually present in adults and associated with less severe outcomes. On the other hand, in 7% of the patients an X-linked inherited genetic defect was reported (mostly in TAZ, causing Barth’s syndrome), a mitochondrial defect was found in 7%, while 6% presented a chromosome defect (frequently 1p36 deletion). These abnormalities were mostly observed in children and were associated with a higher risk for severe outcomes, congenital heart defects (CHD), and neuromuscular symptoms25.
Meanwhile, as in sporadic forms, a pathological genetic defect may be also absent in a considerable number of patients with familial NCCM, indicating that many genetic causes are still unknown4,9. However, a recent retrospective analysis showed that the risk of left ventricular systolic dysfunction (LVSD) was lower in these two scenarios compared to patients with a mutation and that LVSD was associated with cardiac events in mutation carriers but not in sporadic cases7.
Epidemiology
The real incidence and prevalence of NCCM remain unknown3. In adults, the prevalence may vary from 0.14% in the general population up to 3.7% in patients with LVSD26,27. In children, it represents 4.8% to 9% of all cardiomyopathies, the third after DCM and HCM12,28.
Diagnosis
The morphological hallmark of NCCM consists of prominent trabeculae, intratrabecular recesses and a bilayered myocardium composed of a compacted and noncompacted layers2,3. Despite lacking gold standard criteria, the diagnosis mostly relies on non-invasive imaging techniques, usually transthoracic echocardiography (TTE) and cardiac magnetic resonance (CMR)3,4,29. Additionally, there is evidence that these features may represent a remodelling process secondary to abnormal volume/pressure loading conditions, whether physiological and transient (pregnancy, athleticism) or pathological (hypertension, sickle cell disease)7,10,11,29, and that a phenotypic overlapping with other cardiomyopathies may also be seen3,7,9,29. Considering the hypothesis that noncompaction may represent an epiphenomenon under certain conditions, we agree with Negri et al.29 that the fulfilment of imaging criteria should be complemented with clinical (including family history), electrocardiographic, and genetic findings to establish the diagnosis. Accordingly, we propose the use of a multifactorial algorithm to establish the diagnosis of NCCM (Figure 1).
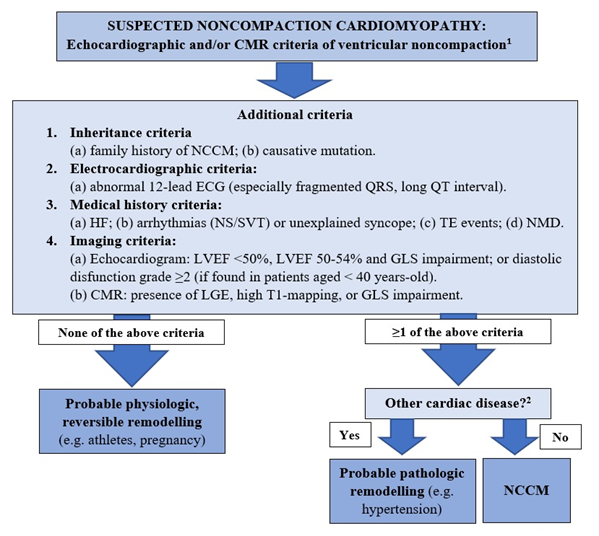
Figure 1: Proposed multifactorial diagnostic algorithm for noncompaction cardiomyopathy.
CMR: cardiac magnetic resonance; ECG: electrocardiogram; GLS: global longitudinal strain; HF: heart failure; LGE: late gadolinium enhancement; LVEF: left ventricular ejection fraction; NMD: neuromuscular disease; NS/SVT: non-sustained or sustained ventricular tachycardia; TE: thromboembolic; 1 Jenni’s criteria are recommended for echocardiography and Jacquier’s are preferred over Peterson’s criteria for CMR; 2 Does not generally apply to congenital heart defects.
This complete evaluation also allows the establishment of eight phenotypical subtypes of NCCM, as initially proposed by Towbin et al.3, each one being associated with different outcomes. A summarized description of the NCCM subtypes, based on simple cardiac imaging measurements (e.g. LV wall size and thickness), co-existing findings such as CHD or arrhythmias, and corresponding prognosis, is described in Table 1.
Table 1: Noncompaction cardiomyopathy phenotypical subtypes and corresponding prognosis*.
|
NCCM subtype |
LV size |
LV wall thickness |
LV systolic function |
LV diastolic function |
RV involvement |
Arrhythmias |
CHD |
Estimated prognosis |
|
Benign |
N |
N |
N |
N |
- |
- |
- |
Good |
|
Dilated LV |
↑ |
↑ / N |
↓ / N |
↓ / N |
+ / - 2 |
+ / - |
- |
Similar to DCM |
|
Hypertrophic LV |
N / ↓ |
↑ |
↑ / N |
↓ |
+ / - 2 |
+ / - |
- |
Good / similar to HCM |
|
Hypertrophic dilated LV |
↑ |
↑ |
↓ |
↓ |
+ / - 2 |
+ / - |
- |
Poor |
|
Restrictive LV |
N |
N / ↑ |
N |
↓ 1 |
+ / - 2 |
+ / - |
- |
Poor |
|
RV or biventricular |
N / ↑ |
N / ↑ |
N / ↓ |
N / ↓ |
+ |
+ / - |
- |
Unknown (for RV involvement) |
|
With arrhythmias |
N |
N |
N |
N / ↓ |
|
+ (VA) |
- |
Worse (versus no VA) |
|
With CHD |
N /↑ |
N / ↑ |
N / ↓ |
N / ↓ |
+ / -2 |
+ / - |
+ |
Depends on CHD type |
CHD: congenital heart disease; DCM: dilated cardiomyopathy; HCM: hypertrophic cardiomyopathy; LV: left ventricle; N: normal; NCCM: noncompaction cardiomyopathy; RV: right ventricle; VA: ventricular arrhythmias; ↑ increase; ↓ decrease;1 atrial dilation;2 secondary to left ventricular dysfunction and/or the co-existent congenital heart defect.
*Content based on Towbin et al.3
Clinical presentation
Typically, NCCM is characterized by the clinical triad of heart failure (HF), arrhythmias and thromboembolic episodes22,23,26,30, with overall reported rates of 9%-89%, 9%-41%, and 4%-7% respectively29. Both HF and ventricular arrhythmias (VA) preclude worse outcome31,32. However, the clinical presentation may be highly variable, ranging from asymptomatic in almost half of the cases7,9,25,29, atypical symptoms like effort angina33,34, to a considerable frequency of sudden cardiac death (SCD)7,25,31,34. Neuromuscular disorders (such as myotonic dystrophies, Barth syndrome, or mitochondrial disorders), and CHD (mostly right-sided defects – Ebstein’s anomaly, pulmonary artery stenosis/atresia, or tricuspid atresia) may be observed, especially in paediatric populations, and usually lead to worse prognosis3,8,35.
Electrocardiogram
Electrocardiographic abnormalities can be seen in more than 80% of cases, most frequently including ventricular hypertrophy voltage criteria, T-wave inversion, ST-segment strain, prolonged QT, fragmented QRS, right/left bundle branch block, and pre-excitation3,22,26. Regular Holter monitoring should be used in symptomatic patients, especially in case of LSVD, to identify clinically relevant arrhythmias24,26.
Cardiac imaging
Transthoracic echocardiography remains the most common and initial diagnostic strategy, mainly due to its widespread availability, ease of interpretability, and low cost3,36. It allows a simple and reliable evaluation of ventricular size and function, wall thickness, and eventually associated CHD. Among the echocardiographic criteria, those proposed by Chin et al.2 and by Jenni et al.37 are the most widely used although the latter is usually preferred given its higher specificity and necropsy validation4,29. Jenni et al.37 consider for the diagnosis of NCCM the presence of a bilayered myocardial structure consisting of a compacted thin epicardial band (C) and a much thicker non-compacted endocardial layer (NC), with a maximal NC/C ratio >2 at end-systole on short-axis parasternal views, in association with deep perfused intertrabecular recesses on colour Doppler. In this study, abnormalities were typically located in the mid-lateral, mid-inferior, and apical LV areas, and patients with CHD were excluded. As stated, the right ventricle may also be involved4,26, although no recognized criteria are available3. Contrast-enhanced TTE may be useful in cases of poor acoustic window and in differentiating NCCM from HCM36. Recently, advanced techniques (strain, strain rate, torsion) are being used3,4 and the presence of base-apex longitudinal strain gradient may assist in differentiating between NCCM and DCM38.
Given the higher quality and sensitivity in the detection of myocardial trabeculations, CMR has an important role in the confirmation of NCCM and also allows the identification/exclusion of thrombi and other coexisting abnormalities4,36. The same controversies remain regarding diagnostic criteria, however, the most used are those of Peterson et al.39, consisting of a NC/C myocardium ratio >2.3 at end-diastole, and Jacquier et al.40 which requires a NC ventricular mass >20% of the global LV mass for diagnosis. Aside from ventricular strain36, CMR also allows tissue characterization using late gadolinium enhancement (LGE) and T1-mapping to identify myocardial fibrosis, which could behave as a prognostic marker4,30. In a multicentre study by Andreini et al.41, LGE (replacement fibrosis), LVSD and LV dilation were independently associated with cardiac events, while the degree of trabeculation was not. Additionally, T1-mapping demonstrates an expansion of the extracellular space by diffuse myocardial fibrosis, normally not identifiable by LGE (less sensitive), which is correlated with myocardial dysfunction and VA42.
Finally, cardiac computed tomography is useful for the anatomical and functional assessment of both ventricles and allows the exclusion of coronary artery disease3,4, which is typically absent3,16.
Genetic testing
Although the diagnostic yield is usually low7,34,43, genetic screening should be offered to (index) patients with an established clinical diagnosis of NCCM44,45. Additionally, recent evidence emphasizes the importance of routine genetic screening given its role in risk stratification and prognosis7,25.
Treatment
There are no specific guidelines for the therapeutic management of NCCM, so it is largely dictated by clinical ï¬ndings3. In case of HF, patients with LSVD should be managed based on existing recommendations - typically including oral angiotensin-converting enzyme inhibitors, β blockers, and aldosterone antagonist46, while treatment of those presenting preserved ejection fraction is more challenging and targeted on symptom relief and comorbidities47. Loop diuretics should be used in case of pulmonary or peripheral congestion or volume overload46,47. Cardiac resynchronisation may be also considered and seems to provide beneficial clinical effects and LV function improvement48. Ventricular assist devices and cardiac transplantation are possibilities for end-stage disease3.
As NCCM carries an increased risk of VA and SCD7,26,30,34, implantable cardioverter defibrillator is recommended in case of ventricular tachyarrhythmias (VT) associated with syncope or SCD and should be implanted in the presence of non-sustained VT and LVSD45.
Despite the association with increased thromboembolic risk23,30, routine use of anticoagulants is still a matter of debate4. While oral anticoagulation is recommended in patients with atrial fibrillation and/or previous embolic event45, it should be used in case of intracardiac thrombi49 and may be reasonable in individuals with LVSD30,45,49. Vitamin-K antagonists have been advocated as a preferred choice due to some concerns regarding non-vitamin-K oral anticoagulants49, although these drugs proved to be safer and equally effective in adults with non-valvular atrial fibrillation50.
Family Screening
Given that up to 50% of cases may be familial9,22,23, mutation-specific testing is recommended for family members and appropriate relatives following the identification of a NCCM-causative mutation in the index case44,45. Regular cardiac follow-up (including TTE and electrocardiogram) is recommended for family members presenting the same mutation and for first-degree relatives of an index case with no mutation or not tested, since childhood up to 60 years-old43.
Conclusions
Almost a century has passed since its first description and NCCM remains a chameleonic entity: from aetiopathogenesis to clinical presentation and prognosis. Recently, the emphasis has been given to genetics and genotype-phenotype relation, with many studies suggesting that noncompaction may be found in a considerable proportion of patients without causative mutation or familial history (sporadic) as a consequence of ventricular remodelling due to certain physiological/pathologic overload conditions. These patients usually present a good prognosis, contrary to those with an identified genetic abnormality. Additionally, advanced imaging techniques (LGE and T1-mapping) have demonstrated an increasingly important role in risk stratification. However, these and many other aspects are still not fully understood and multicentre registries/studies are required to assess the utility of diagnostic criteria, provide risk stratification tools, and determine the appropriate management/treatment.
References
- Grant RT. An unusual anomaly of the coronary vessels in the malformed heart of a child. Heart. 1926; 13: 272–83.
- Chin TK, Perloff JK, Williams RG, et al. Isolated noncompaction of left ventricular myocardium. A study of eight cases. Circulation. 1990; 82: 507–13.
- Towbin JA, Lorts A, Jefferies JL. Left ventricular non-compaction cardiomyopathy. Lancet. 2015; 386: 813–25.
- Lorca R, Rozado J, Martín M. Non Compaction cardiomyopathy: Review of a controversial entity. Med Clin (Barc). 2018; 150: 354–60.
- Maron BJ, Towbin JA, Thiene G, et al. Contemporary Definitions and Classification of the Cardiomyopathies: An American Heart Association Scientific Statement From the Council on Clinical Cardiology, Heart Failure and Transplantation Committee; Quality of Care and Outcomes Research and Functional Genomics and Translational Biology Interdisciplinary Working Groups; and Council on Epidemiology and Prevention. Circulation. 2006; 113: 1807–16.
- Elliott P, Andersson B, Arbustini E, et al. Classification of the cardiomyopathies: a position statement from the european society of cardiology working group on myocardial and pericardial diseases. Eur Heart J. 2007; 29: 270–6.
- van Waning JI, Caliskan K, Hoedemaekers YM, et al. Genetics, Clinical Features, and Long-Term Outcome of Noncompaction Cardiomyopathy. J Am Coll Cardiol. 2018; 71: 711–22.
- Ting TW, Jamuar SS, Brett MS, et al. Left Ventricular Non-compaction: Is It Genetic? Pediatr Cardiol. 2015; 36: 1565–72.
- van Waning JI, Caliskan K, Michels M, et al. Cardiac Phenotypes, Genetics, and Risks in Familial Noncompaction Cardiomyopathy. J Am Coll Cardiol. 2019; 73: 1601–11.
- Gati S, Chandra N, Bennett RL, et al. Increased left ventricular trabeculation in highly trained athletes: do we need more stringent criteria for the diagnosis of left ventricular non-compaction in athletes? Heart. 2013; 99: 401–8.
- Gati S, Papadakis M, Papamichael ND, et al. Reversible De Novo Left Ventricular Trabeculations in Pregnant Women: Implications for the Diagnosis of Left Ventricular Noncompaction in Low-Risk Populations. Circulation. 2014; 130: 475–83.
- Lee TM, Hsu DT, Kantor P, et al. Pediatric Cardiomyopathies. Circ Res. 2017; 121: 855–73.
- Henderson DJ, Anderson RH. The Development and Structure of the Ventricles in the Human Heart. Pediatr Cardiol. 2009; 30: 588–96.
- Sedmera D, Pexieder T, Vuillemin M, et al. Developmental patterning of the myocardium. Anat Rec. 2000; 258: 319–37.
- Chen H, Zhang W, Li D, et al. Analysis of Ventricular Hypertrabeculation and Noncompaction Using Genetically Engineered Mouse Models. Pediatr Cardiol. 2009; 30: 626–34.
- Ichida F. Left ventricular noncompaction − Risk stratification and genetic consideration. J Cardiol. 2020; 75: 1–9.
- Luxan G, Casanova JC, Martínez-Poveda B, et al. Mutations in the NOTCH pathway regulator MIB1 cause left ventricular noncompaction cardiomyopathy. Nat Med. 2013; 19: 193–201.
- Stollberger C, Finsterer J, Blazek G. Left ventricular hypertrabeculation/noncompaction and association with additional cardiac abnormalities and neuromuscular disorders. Am J Cardiol. 2002; 90: 899–902.
- Finsterer J, Stollberger C, Towbin JA. Left ventricular noncompaction cardiomyopathy: cardiac, neuromuscular, and genetic factors. Nat Rev Cardiol. 2017; 14: 224–37.
- Wengrofsky P, Armenia C, Oleszak F, et al. Left Ventricular Trabeculation and Noncompaction Cardiomyopathy: A Review. EC Clin Exp Anat. 2019; 2: 267–83.
- Finsterer J, Stollberger C, Schubert B. Acquired left ventricular noncompaction as a cardiac manifestation of neuromuscular disorders. Scand Cardiovasc J. 2008; 42: 25–30.
- Ichida F, Hamamichi Y, Miyawaki T, et al. Clinical features of isolated noncompaction of the ventricular myocardium. J Am Coll Cardiol. 1999; 34: 233–40.
- Oechslin EN, Attenhofer Jost CH, Rojas JR, et al. Long-term follow-up of 34 adults with isolated left ventricular noncompaction: a distinct cardiomyopathy with poor prognosis. J Am Coll Cardiol. 2000; 36: 493–500.
- Sedaghat-Hamedani F, Haas J, Zhu F, et al. Clinical genetics and outcome of left ventricular non-compaction cardiomyopathy. Eur Heart J. 2017; 38: 3449–60.
- van Waning JI, Moesker J, Heijsman D, et al. Systematic Review of Genotype-Phenotype Correlations in Noncompaction Cardiomyopathy. J Am Heart Assoc. 2019; 8: e012993.
- Aras D, Tufekcioglu O, Ergun K, et al. Clinical Features of Isolated Ventricular Noncompaction in Adults Long-Term Clinical Course, Echocardiographic Properties, and Predictors of Left Ventricular Failure. J Card Fail. 2006; 12: 726–33.
- Sandhu R, Finkelhor RS, Gunawardena DR, et al. Prevalence and Characteristics of Left Ventricular Noncompaction in a Community Hospital Cohort of Patients with Systolic Dysfunction. Echocardiography. 2008; 25: 8-12.
- Nugent AW, Daubeney PEF, Chondros P, et al. The Epidemiology of Childhood Cardiomyopathy in Australia. N Engl J Med. 2003; 348: 1639–46.
- Negri F, De Luca A, Fabris E, et al. Left ventricular noncompaction, morphological, and clinical features for an integrated diagnosis. Heart Fail Rev. 2019; 24: 315–23.
- Ikeda U, Minamisawa M, Koyama J. Isolated left ventricular non-compaction cardiomyopathy in adults. J Cardiol. 2015; 65: 91–7.
- Brescia ST, Rossano JW, Pignatelli R, et al. Mortality and Sudden Death in Pediatric Left Ventricular Noncompaction in a Tertiary Referral Center. Circulation. 2013; 127: 2202–8.
- Tollberger C, Blazek G, Gessner M, et al. Neuromuscular comorbidity, heart failure, and atrial fibrillation as prognostic factors in left ventricular hypertrabeculation/noncompaction. Herz. 2015; 40: 906–11.
- Santos LG, Carvalho R, Montenegro F, et al. Left Ventricular Noncompaction: A Rare Cause of Effort Angina Unveiled During Left Heart Catheterization. J Invasive Cardiol. 2020; 32: E42.
- Miller EM, Hinton RB, Czosek R, et al. Genetic Testing in Pediatric Left Ventricular Noncompaction. Circ Cardiovasc Genet. 2017; 10: e001735.
- Stollberger C, Wegner C, Finsterer J. Left ventricular hypertrabeculation/noncompaction, cardiac phenotype, and neuromuscular disorders. Herz. 2019; 44: 659–65.
- Mavrogeni SI, Markousis-Mavrogenis G, Vartela V, et al. The pivotal role of cardiovascular imaging in the identification and risk stratification of non-compaction cardiomyopathy patients. Heart Fail Rev. 2019; doi: 10.1007/s10741-019-09898-8. [online ahead of print].
- Jenni R. Echocardiographic and pathoanatomical characteristics of isolated left ventricular non-compaction: a step towards classification as a distinct cardiomyopathy. Heart. 2001; 86: 666–71.
- Tarando F, Coisne D, Galli E, et al. Left ventricular non-compaction and idiopathic dilated cardiomyopathy: the significant diagnostic value of longitudinal strain. Int J Cardiovasc Imaging. 2017; 33: 83–95.
- Petersen SE, Selvanayagam JB, Wiesmann F, et al. Left Ventricular Non-Compaction. J Am Coll Cardiol. 2005; 46: 101–5.
- Jacquier A, Thuny F, Jop B, et al. Measurement of trabeculated left ventricular mass using cardiac magnetic resonance imaging in the diagnosis of left ventricular non-compaction. Eur Heart J. 2010; 31: 1098–104.
- Andreini D, Pontone G, Bogaert J, et al. Long-Term Prognostic Value of Cardiac Magnetic Resonance in Left Ventricle Noncompaction. J Am Coll Cardiol. 2016; 68: 2166–81.
- Araujo-Filho JAB, Assuncao AN, Tavares de Melo MD, et al. Myocardial T1 mapping and extracellular volume quantification in patients with left ventricular non-compaction cardiomyopathy. Eur Heart J Cardiovasc Imaging. 2018; 19: 888–95.
- Charron P, Arad M, Arbustini E, et al. Genetic counselling and testing in cardiomyopathies: a position statement of the European Society of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur Heart J. 2010; 31: 2715–26.
- Ackerman MJ, Priori SG, Willems S, et al. HRS/EHRA Expert Consensus Statement on the State of Genetic Testing for the Channelopathies and Cardiomyopathies. Heart Rhythm. 2011; 8: 1308–39.
- Towbin JA, McKenna WJ, Abrams DJ, et al. 2019 HRS expert consensus statement on evaluation, risk stratification, and management of arrhythmogenic cardiomyopathy. Heart Rhythm. 2019; 16: e301–72.
- Ponikowski P, Voors AA, Anker SD, et al. 2016 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure: The Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC) Developed with the special contribution of the Heart Failure Association (HFA) of the ESC. Eur Heart J. 2016; 37: 2129–200.
- Lopes Fernandes S, Ribeiro Carvalho R, Graça Santos L, et al. Pathophysiology and Treatment of Heart Failure With Preserved Ejection Fraction: State of the Art and Prospects for the Future. Arq Bras Cardiol. 2019; 114: 120-9.
- Bertini M, Balla C, Pavasini R, et al. Efficacy of cardiac resynchronization therapy in patients with isolated ventricular noncompaction with dilated cardiomyopathy: a systematic review of the literature. J Cardiovasc Med (Hagerstown). 2018; 19: 324–8.
- Finsterer J, Stöllberger C. Primary prophylactic anticoagulation is mandatory if noncompaction is associated with atrial fibrillation or heart failure. Int J Cardiol. 2015; 184: 268–9.
- Ruff CT, Giugliano RP, Braunwald E, et al. Comparison of the efficacy and safety of new oral anticoagulants with warfarin in patients with atrial fibrillation: a meta-analysis of randomised trials. Lancet. 2014; 383: 955-962.